🤩 WGCNA | 值得你深入学习的生信分析方法!~(网状分析-第二步补充-大数据的网络构建与模块识别)

🤩 WGCNA | 值得你深入学习的生信分析方法!~(网状分析-第二步补充-大数据的网络构建与模块识别)

生信漫卷
发布于 2023-02-24 14:30:20
发布于 2023-02-24 14:30:20
1写在前面
之前我们完成了WGCNA输入数据的清洗,网络构建和模块识别。😘
但这里我们的示例数据内所含有的基因其实是很少的,而在实际情况中,一个简单的测序可能就要包含上万个基因,这对大家的电脑无疑是不小的压力。🤒
在WGCNA的包内其实也提供了解决方案,基本思想是分级聚类。🧐
1️⃣ 首先,我们使用快速但相对粗糙的聚类方法,用于将基因预聚类成大小接近的模块,且不超过你所设定的基因最大值。😂
2️⃣ 然后我们分别在每个模块中执行完整的网络分析。🤠
3️⃣ 最后,合并特征基因高度相关的模块。😏
2用到的包
rm(list = ls())
library(WGCNA)
library(tidyverse)
3示例数据
load("FemaleLiver-01-dataInput.RData")
4软阈值
4.1 topology analysis
首先我们还是要和之前一样进行soft thresholding power β的计算。🤒
powers <- c(c(1:10), seq(from = 12, to=20, by=2))
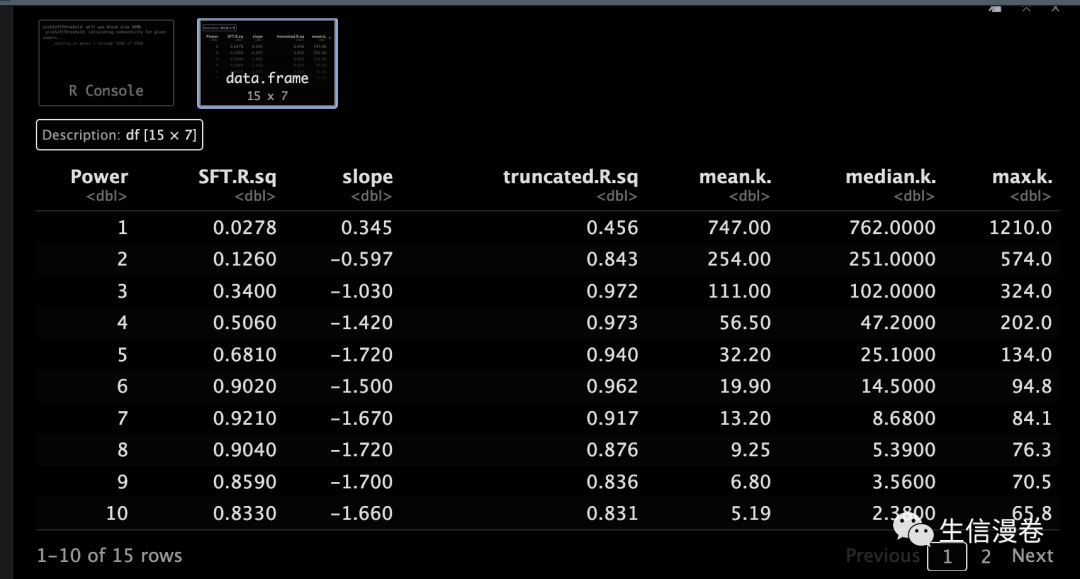
sft <- pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
4.2 可视化
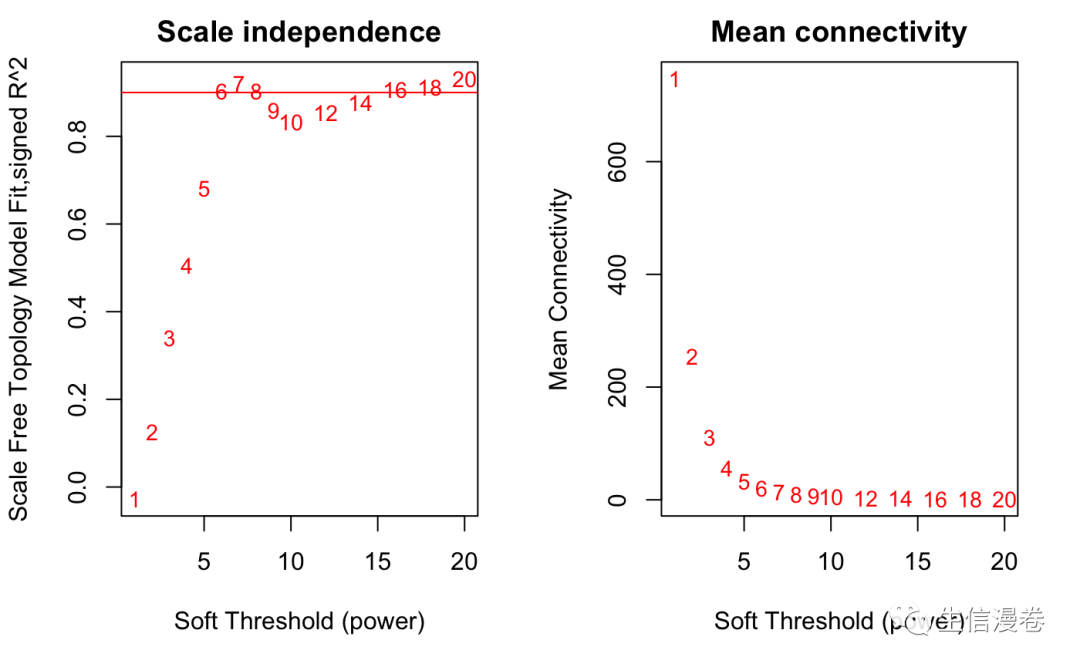
显然,我们的结果和之前是一样的,6。😜
sizeGrWindow(9, 5)
par(mfrow = c(1,2))
cex1 = 0.9
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab = "Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",
type="n", main = paste("Scale independence"))
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red")
abline(h=0.90,col="red")
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity",
type="n", main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
5构建网络与模块识别
5.1 网络构建
这里我们就要设置每个block的最大size是多少了,分次计算,再合并,以此减少内存的负担。🤒
bwnet <- blockwiseModules(
datExpr, maxBlockSize = 2000,
power = 6, TOMType = "unsigned", minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = T,
saveTOMs = T,
saveTOMFileBase = "femaleMouseTOM-blockwise",
verbose = 3)
5.2 查看模块数
table(bwnet$colors)

6对比结果
这里我们再对比一下结果,看看2种方法得出结果的区别。🤪
6.1 载入之前的结果
这里我们把之前不分次计算的结果载入进来,后面会用到label和colors。😷
load(file = "FemaleLiver-02-networkConstruction-auto.RData")
6.2 匹配颜色
把colors和label匹配起来,嘿嘿。🤨
bwLabels <- matchLabels(bwnet$colors, moduleLabels);
bwModuleColors <- labels2colors(bwLabels)
6.3 分次结果可视化
sizeGrWindow(6,6)
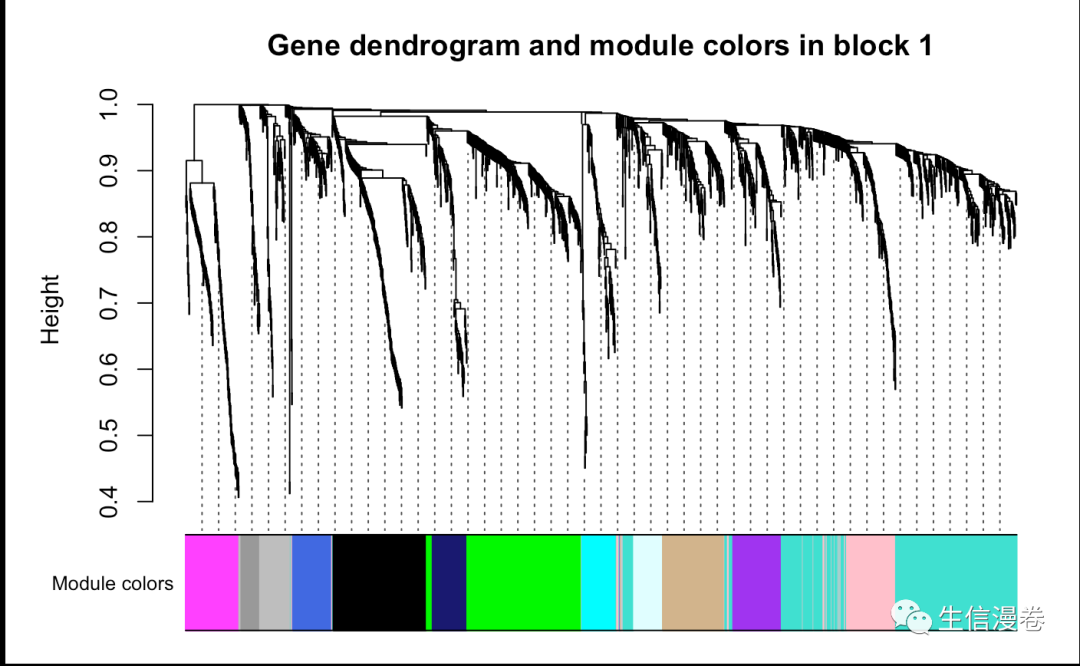
# Block 1
plotDendroAndColors(bwnet$dendrograms[[1]], bwModuleColors[bwnet$blockGenes[[1]]],
"Module colors", main = "Gene dendrogram and module colors in block 1",
dendroLabels = F, hang = 0.03,
addGuide = T, guideHang = 0.05)
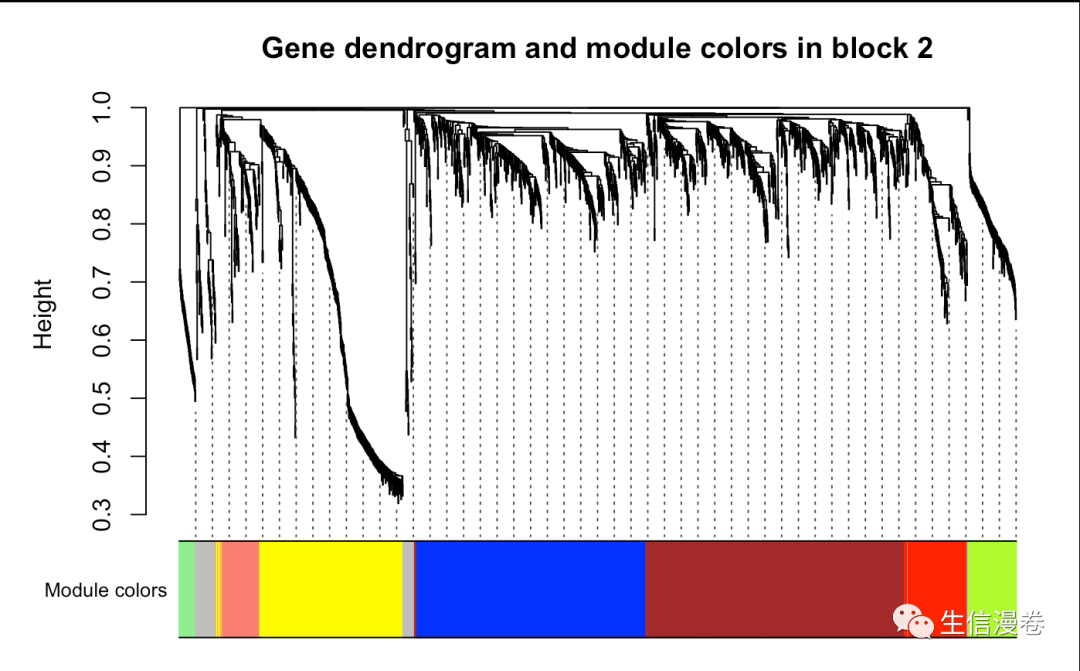
# Block 2
plotDendroAndColors(bwnet$dendrograms[[2]], bwModuleColors[bwnet$blockGenes[[2]]],
"Module colors", main = "Gene dendrogram and module colors in block 2",
dendroLabels = F, hang = 0.03,
addGuide = T, guideHang = 0.05)
7对比两种方法的结果差异
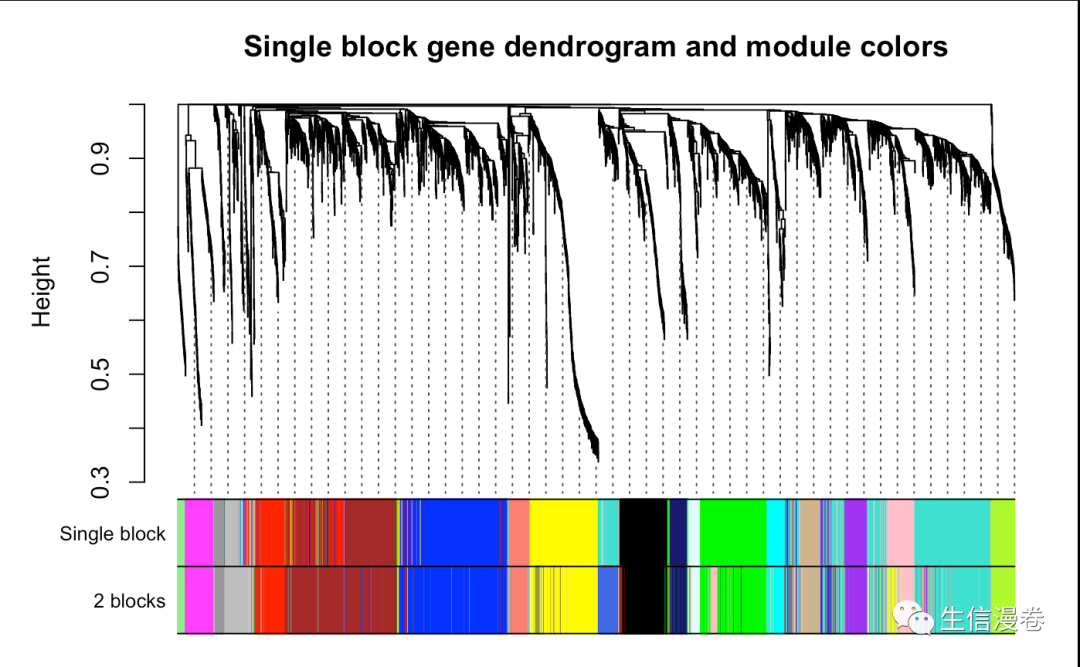
7.1 对比一下
我们以可视化的形式对比一下,分割出来的模块差异不大。😂
sizeGrWindow(12,9)
plotDendroAndColors(geneTree,
cbind(moduleColors, bwModuleColors),
c("Single block", "2 blocks"),
main = "Single block gene dendrogram and module colors",
dendroLabels = F, hang = 0.03,
addGuide = T, guideHang = 0.05)
7.2 对比eigengenes
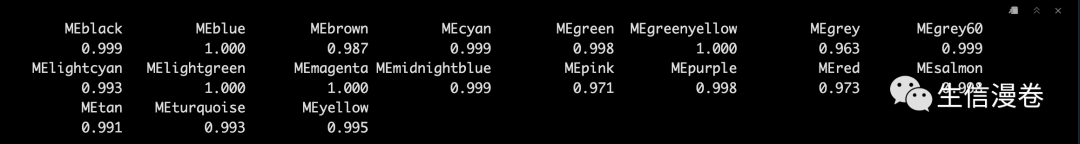
这里我们提取一下2种方法得到的module eigengenes。🥳
singleBlockMEs <- moduleEigengenes(datExpr, moduleColors)$eigengenes
blockwiseMEs <- moduleEigengenes(datExpr, bwModuleColors)$eigengenes
match之后看一下结果,嘿嘿。🫶
高度一致,所以这种blockwise的方法,请放心食用吧,各位。😉
single2blockwise <- match(names(singleBlockMEs), names(blockwiseMEs))
signif(diag(cor(blockwiseMEs[, single2blockwise], singleBlockMEs)), 3)
8如何引用
📍
Langfelder, P., Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). https://doi.org/10.1186/1471-2105-9-559
最后祝大家早日不卷!~
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-02-11,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号