学习小组Day6-bubble
原创

学习R包
R包是多个函数的集合,具有详细的说明和示例。
学生信,R语言必学的原因是丰富的图表和Biocductor上面的各种生信分析R包。 包的使用是一通百通的。
1.安装并加载R包
1.1 镜像设置
也和Linux一样,官方源因受到网速影响比较慢,添加国内镜像源会方便很多
这里需要用到两行代码
# options函数就是设置R运行过程中的一些选项设置
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")) #对应清华源
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/") #对应中科大源
# 当然可以换成其他地区的镜像
options()$BioC_mirro #检验默认镜像
options()$reposr # 查询自己的镜像这种是每一次打开都要重新设置一次的
还有一种像Linux一样直接修改R中的相当于Linux中的.bashrc/环境文件一样的R的环境文件.Rprofile即可
首先用file.edit()来编辑文件:
file.edit('~/.Rprofile')然后在文件中添加上述两行代码即可保存重新加载一下R(很像Linux中的source ~/.bashrc)
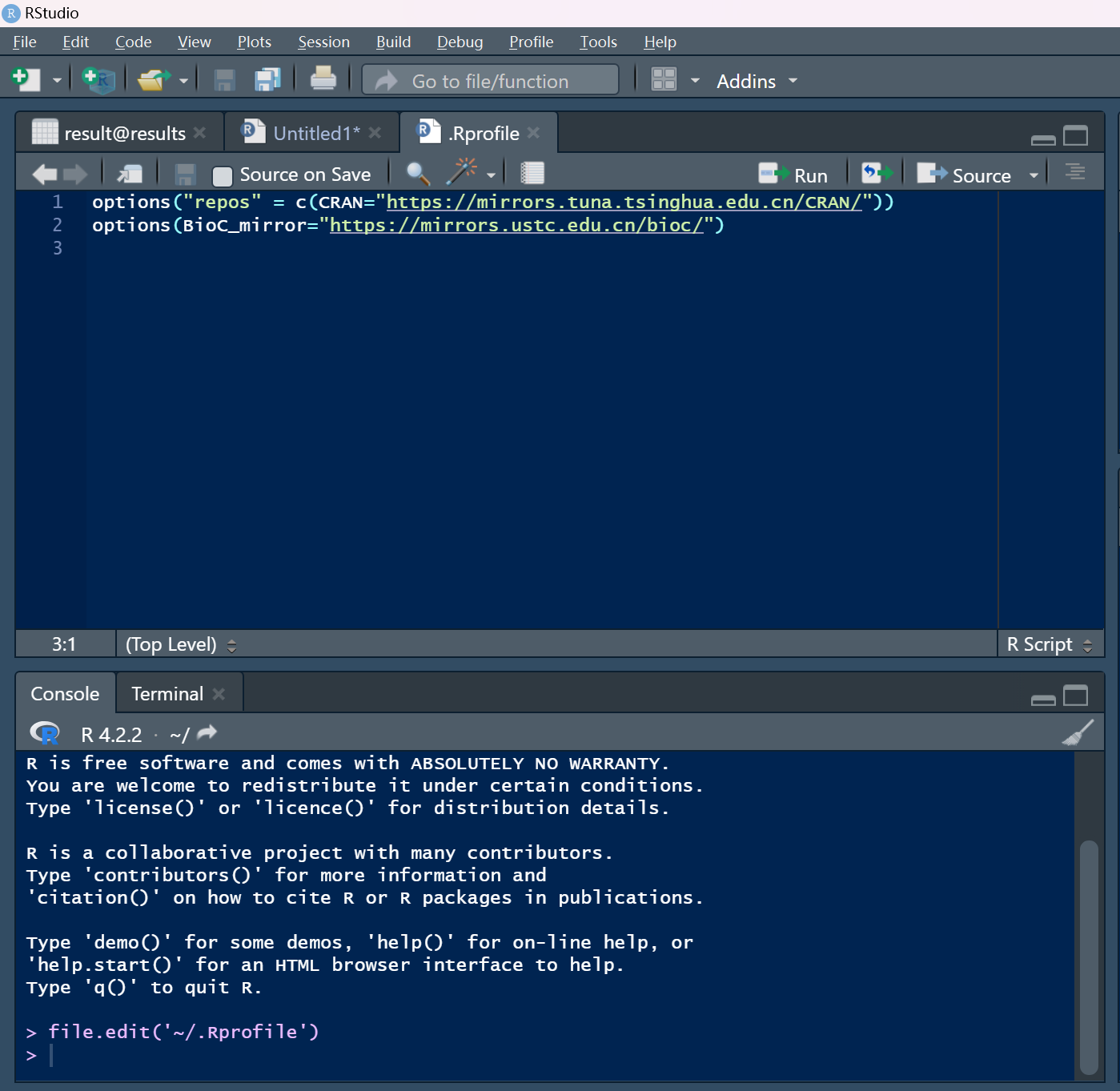
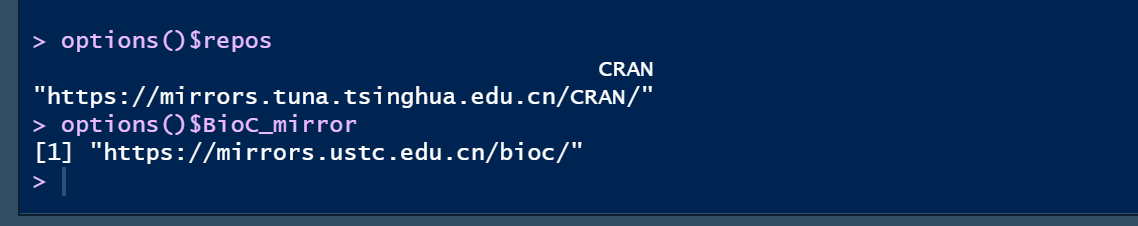
可以看到配置好镜像啦
1.2 安装
install.packages(“包”) # 普通安装(从CRAN安装)
BiocManager::install(“包”) # 从Bioconductor安装(主要是一些生物学包)
取决于你要安装的包存在于CRAN网站还是Biocductor,存在于哪里?可以谷歌搜到。更多安装来源可以参考
1.3 加载
library和require,两个函数均可。使用一个包,是需要先安装再加载,才能使用包里的函数。
# 安装加载三部曲
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
# 这里是没有修改环境文件的话每次下载记得重新配置
install.packages("dplyr")
library(dplyr)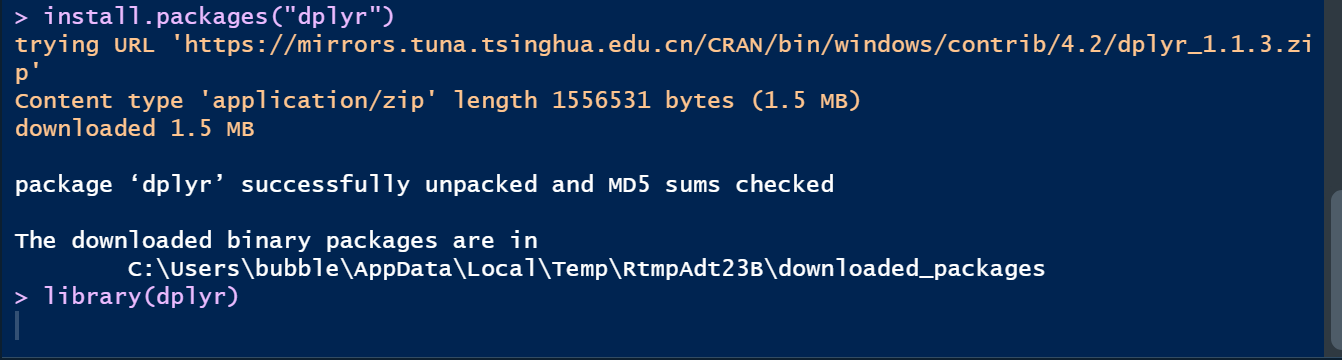
示例数据直接使用内置数据集iris的简化版:
test <- iris[c(1:2,51:52,101:102),]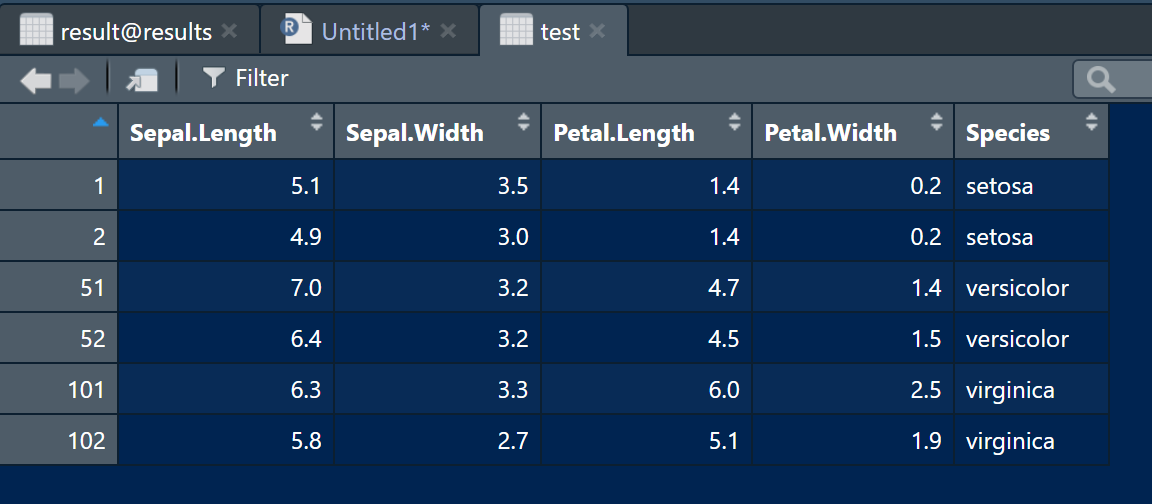
2. 具体讲解dplyr包的五个基础函数
2.1 mutate(),新增列
mutate(test, new = Sepal.Length * Sepal.Width)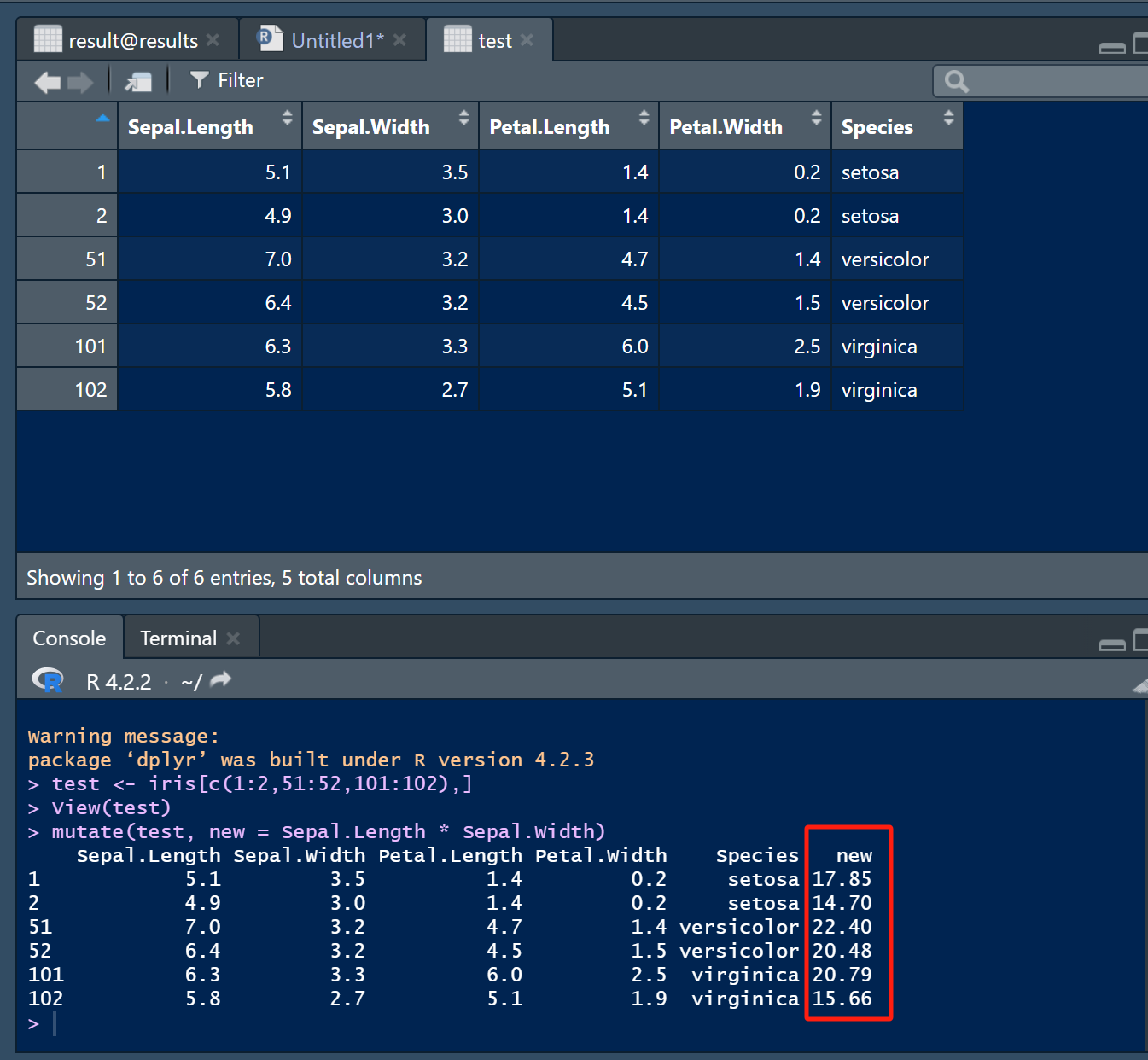
2.2 select(),按列筛选
① 按列号筛选
select(test,1) # 选择第一列
select(test,c(1,5)) # 选择第一和五列
select(test,Sepal.Length) # 直接选择列名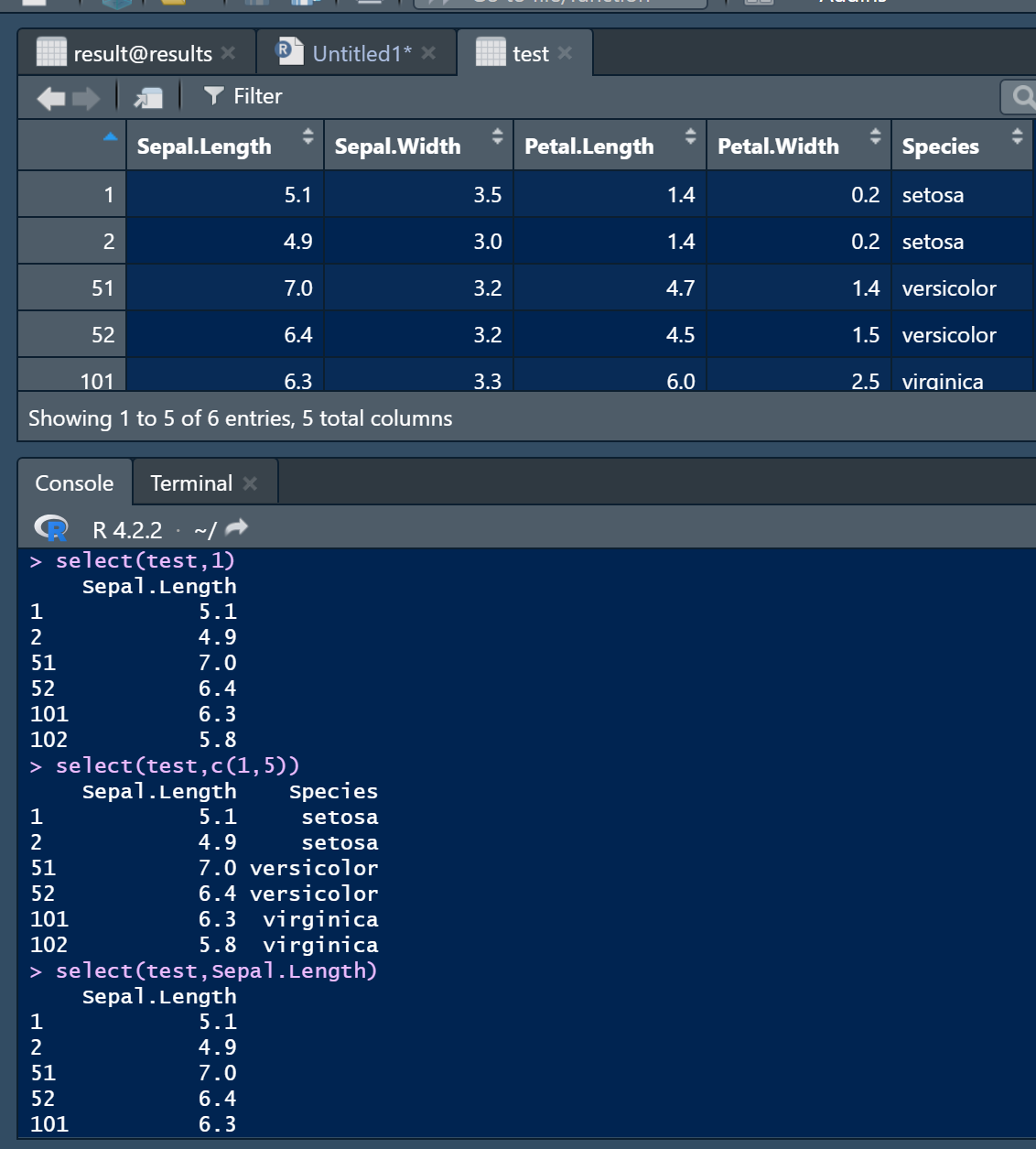
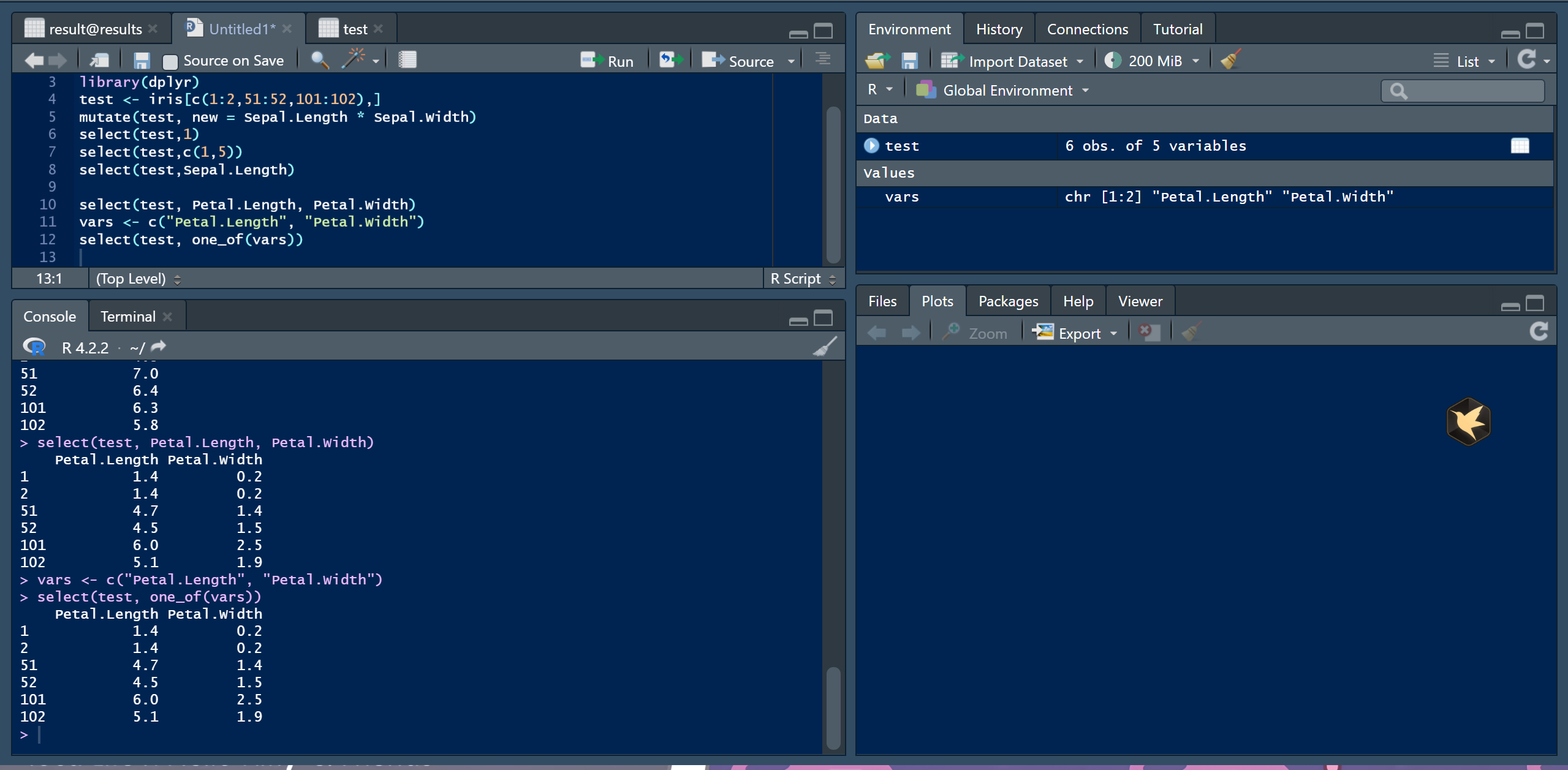
② 按列名筛选
select(test, Petal.Length, Petal.Width)
vars <- c("Petal.Length", "Petal.Width")
select(test, one_of(vars))https://wenku.csdn.net/answer/45me6e3f3a

③ filter()筛选行
filter(test, Species == "setosa")
filter(test, Species == "setosa"&Sepal.Length > 5 )
filter(test, Species %in% c("setosa","versicolor"))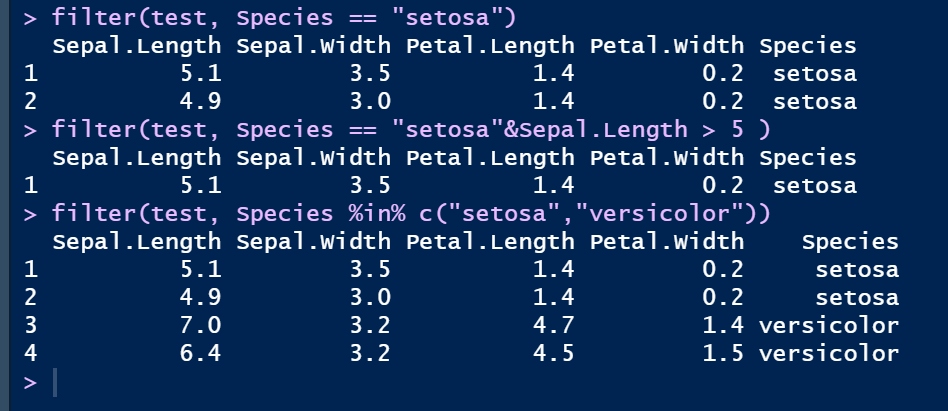
④ arrange(),按某1列或某几列对整个表格进行排序
arrange(test, Sepal.Length)#默认从小到大排序
arrange(test, desc(Sepal.Length))#用desc从大到小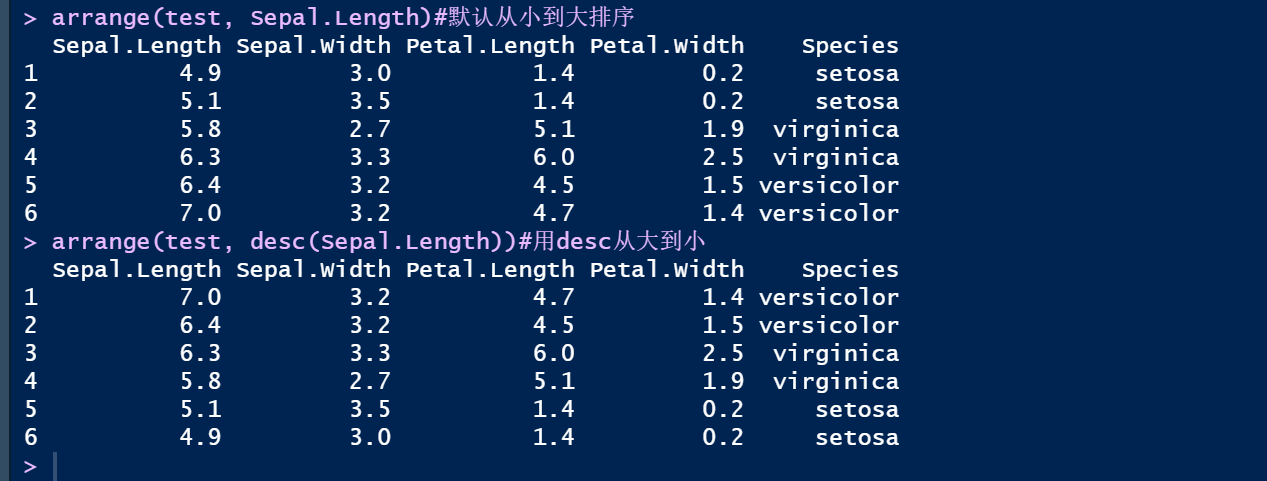
⑤ .summarise():汇总
对数据进行汇总操作,结合group_by使用实用性强
summarise(test, mean(Sepal.Length), sd(Sepal.Length))# 计算Sepal.Length的平均值和标准差
# 先按照Species分组,计算每组Sepal.Length的平均值和标准差
group_by(test, Species)
summarise(group_by(test, Species),mean(Sepal.Length), sd(Sepal.Length))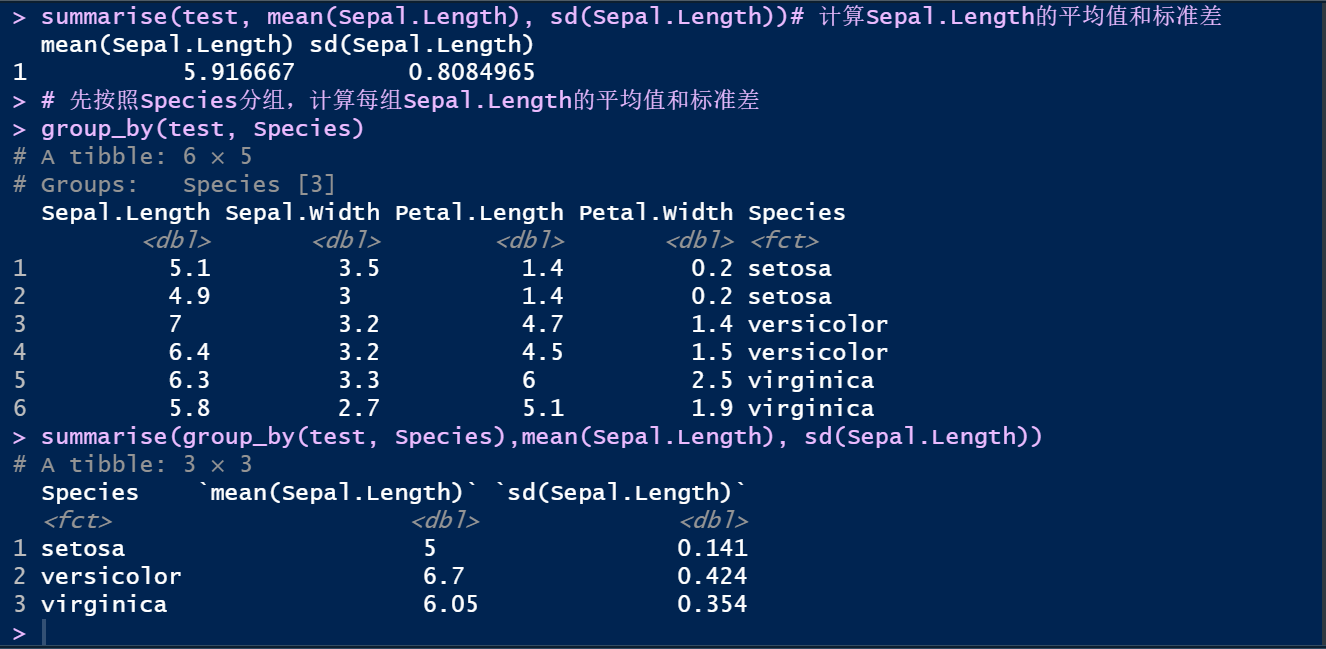
2.3 dplyr两个实用技能
① 管道操作 %>% (cmd/ctr + shift + M)
test %>%
group_by(Species) %>%
summarise(mean(Sepal.Length), sd(Sepal.Length))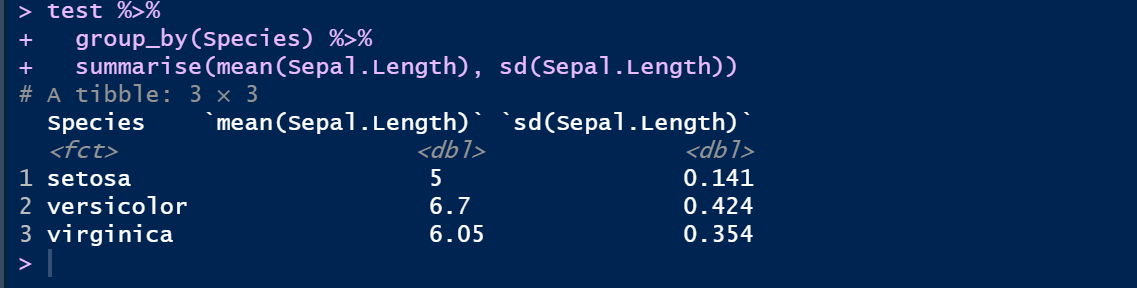
② count统计某列的unique值
count(test,Species)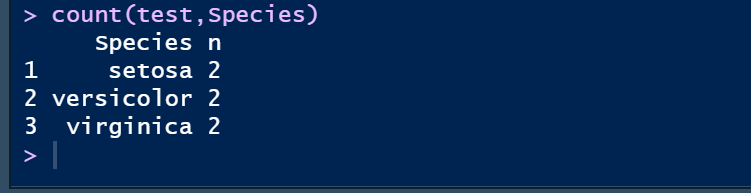
2.4 dplyr处理关系数据
即将2个表进行连接
test1 <- data.frame(x = c('b','e','f','x'),
z = c("A","B","C",'D'))test2 <- data.frame(x = c('a','b','c','d','e','f'),
y = c(1,2,3,4,5,6))先将test1 test2赋值
① 內连inner_join,取交集
inner_join(test1, test2, by = "x")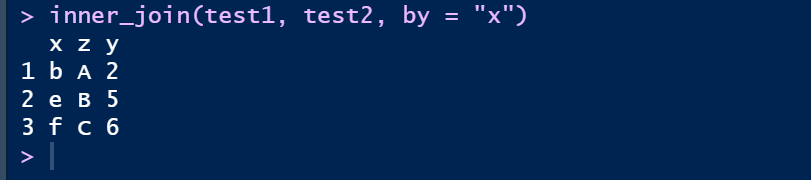
② 左连left_join
left_join(test1, test2, by = 'x')
left_join(test2, test1, by = 'x')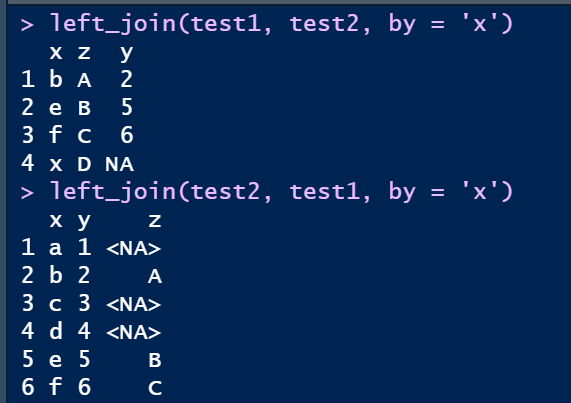
③ 全连full_join
full_join( test1, test2, by = 'x')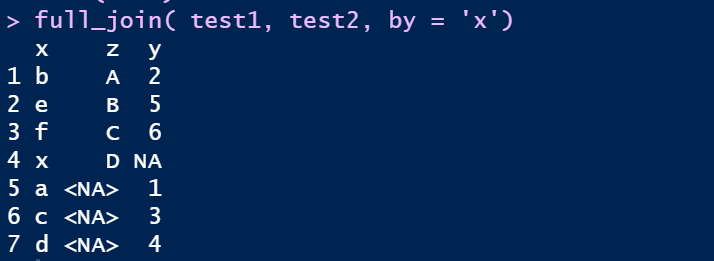
④ 半连接:返回能够与y表匹配的x表所有记录semi_join
semi_join(x = test1, y = test2, by = 'x')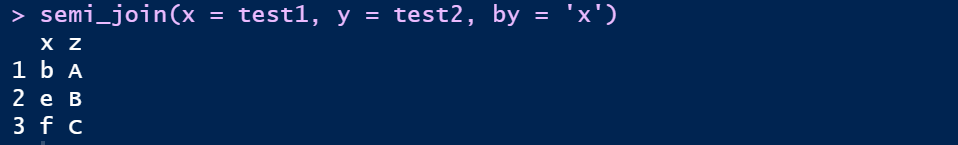
⑤ 反连接:返回无法与y表匹配的x表的所记录anti_join
anti_join(x = test2, y = test1, by = 'x')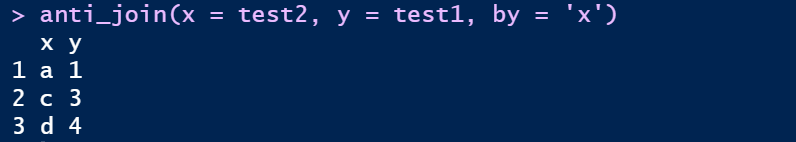
⑥ 简单合并
在相当于base包里的cbind()函数和rbind()函数;注意,bind_rows()函数需要两个表格列数相同,而bind_cols()函数则需要两个数据框有相同的行数
test1 <- data.frame(x = c(1,2,3,4), y = c(10,20,30,40))
test2 <- data.frame(x = c(5,6), y = c(50,60))
test3 <- data.frame(z = c(100,200,300,400))
bind_rows(test1, test2)
bind_cols(test1, test3)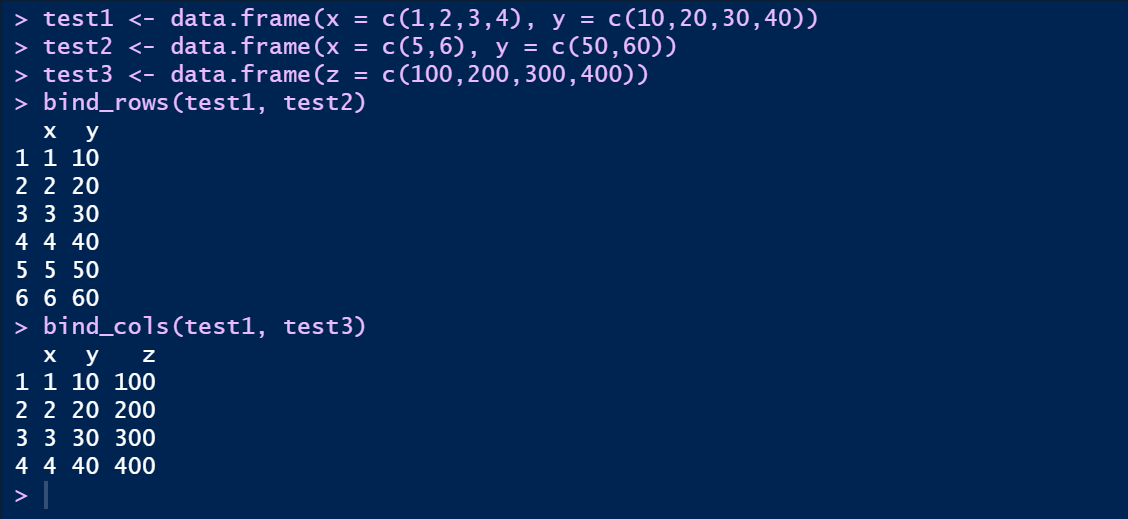
好啦今天的R包学习完啦
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号