蛋白质组学上游数据处理知多少


上游数据分析里面的蛋白质鉴定与定量是两个不同的过程,但是在实践中,蛋白质鉴定和定量通常是联合进行的。例如,在质谱实验中,首先通过MS/MS数据进行蛋白质鉴定,然后利用相应的肽段信号强度或特定的定量标签来量化蛋白质的表达水平。这种结合使用鉴定和定量的方法可以提供关于蛋白质在不同条件下表达模式的详细信息,从而有助于理解生物学过程和疾病机制。
- 蛋白质鉴定(Protein Identification):
- 目的是确定样品中存在的蛋白质的身份。这通常涉及将质谱(MS)产生的肽段的质量和序列信息与数据库中的已知蛋白质序列进行比对。
- 鉴定过程依赖于质谱数据中的肽段指纹图谱或肽段序列的串联质谱(MS/MS)数据,通过数据库搜索来匹配候选蛋白质。
- 蛋白质鉴定可以揭示样品中存在的特定蛋白质或蛋白质的特定形式(如翻译后修饰的蛋白质)。
- 蛋白质定量(Protein Quantitation):
- 目的是测量样品中蛋白质的相对或绝对丰度。定量可以是定性的(比较不同样品中蛋白质的相对表达水平)或绝对定量的(测量蛋白质的拷贝数或浓度)。
- 定量方法包括使用稳定同位素标记的标签(如iTRAQ、TMT、SILAC)或无标记方法(如基于肽段或蛋白质离子强度的LFQ或基于标签的定量)。
- 定量数据可以用于比较不同生物学状态下的蛋白质表达变化,如健康与疾病状态,或响应不同实验处理的条件。
对大家来说,耳熟能详的软件就是 MaxQuant 啦,在综述文章《Bioinformatics Methods for Mass Spectrometry-Based Proteomics Data Analysis》有详细的介绍各种软件的对比汇总。
因为蛋白质搜库软件实在是太多了,包括 MZmine2、MaxQuant、OMSSA、OpenMS、Comet、SuperHirn、MS-GF+、MSFragger、InsPecT、MyriMatch、SpectralST、MS Amanda、Census、Protein Pilot、和msInspect,我们就简单的罗列一些公共数据集里面的常用软件。
基于Mascot
2023年4月的文章:《Targeting ULK1 Decreases IFNγ-Mediated Resistance to Immune Checkpoint Inhibitors 》
提到了:Mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the project accession #: PXD035347.
详见:https://www.ebi.ac.uk/pride/archive/projects/PXD035347
如下所示的描述:
Data were searched using Mascot 2.5 (Matrix Science, http://www.matrixscience.com/index.html) against the human SwissProt database (https://www.uniprot.org/uniprot/?query=reviewed:yes) to identify the proteins processed by nLC-MS/MS, and results were reported at 1% false-discovery rate in Scaffold 4 (Proteome Software).
2024年文章:《Microenvironment Shapes Small Cell Lung Cancer Neuroendocrine States and Presents Unique Therapeutic Opportunities》
详见:https://www.ebi.ac.uk/pride/archive/projects/PXD052033
Peptide identifications, normalization, and log2-transformation for generation of protein-level quantitative data were performed. Briefly, raw data files were searched using Mascot (Matrix Science) and Proteome Discoverer (ThermoFisher Scientific, Inc).
2024的文章:《Serum proteomic for pulmonary infection after pediatric congenital heart surgery》,样品数量,病人队列都不小哦。
To explore serum proteins associated with postoperative pulmonary infection in children with CHD, a total of 98 serum samples collected from the 23 patients before and after surgery were subjected to proteomic analysis
详见:https://www.ebi.ac.uk/pride/archive/projects/PXD051904
Raw data were processed using Proteome Discoverer 2.4 (Thermo Scientific, Waltham, MA) against a human protein database, SwissProt (last modified on June 5, 2023).
基于MaxQuant
MaxQuant的使用方法可以参考这个推文:蛋白组分析软件MaxQuant——Label-free quantification分析教程 有很多蛋白质组数据的作者都是采用了MaxQuant软件,也可以看生信技能树公众号2019的系列推文:
详见:https://www.ebi.ac.uk/pride/archive/projects/PXD051216
Peptides and proteins were identified and quantified using MaxQuant (version 1.6.0.17) searching in Uniprot databases (Homo sapiens and Hepatitis B virus taxonomies, 20210628 download) and in the database of frequently observed contaminants embedded in MaxQuant. Trypsin/P was chosen as the enzyme and 2 missed cleavages were allowed.
详见:https://www.ebi.ac.uk/pride/archive/projects/PXD051130
The resulting MS/MS data were processed using MaxQuant search engine (v.1.6.15.0). Tandem mass spectra were searched against the human SwissProt database (20422 entries) concatenated with reverse decoy database.
值得一提的是很多人会选择Perseus软件,它可以作为MaxQuant的兄弟软件,使用MaxQuant处理之后的数据可以直接导入Perseus进行分析,此软件将处理数据的过程可视化,使用起来简洁明了,并且比在excel中直接分析更加准确。当然了,我们作为生信工程师其实肯定是使用r编程语言来处理蛋白质表达量矩阵啦,并不需要Excel也不需要Perseus软件。
Peaks Xpro software
详见:https://www.ebi.ac.uk/pride/archive/projects/PXD027719
Protein identification was performed in Peaks Xpro software using human protein SwissProt database (https://www.uniprot.org/; organism: Human [9606]; uploaded 02.03.2021; 20394 sequences) and protein contaminants database CRAP (ftp://ftp.thegpm.org/fasta/cRAP; version of 2019-03-04). The search parameters were: parent mass error tolerance 15 ppm and fragment mass error tolerance 0.05 ppm, protein
MS‐GF+ search engine
详见:https://www.ebi.ac.uk/pride/archive/projects/PXD041077
LC–MS/MS data analysis For each MS/MS dataset, post experiment monoisotopic mass refinement (PE‐MMR) was used to accurately assign precursor masses to MS/MS spectra. Resulting MS/MS spectra (i.e., mgf files) were subjected to a database search using the MS‐GF+ search engine (v2017.01.13) in a target decoy setting against the UniProt Human reference database (69,391 entries; February 2016) with the.
详见:https://www.ebi.ac.uk/pride/archive/projects/PXD035504
- Peptide library generation: Individually acquired DIA files were processed using the Signal Extraction module of the DIA-Umpire software tool (DIAu-SE).
- A protein sequence database was built by concatenating the target SwissProt mouse proteome database (canonical sequences), Biognosys iRT peptides for retention time alignment (Biognosys, Schlieren, Switzerland),
每个样品是3个文件:https://ftp.pride.ebi.ac.uk/pride/data/archive/2023/03/PXD035504/
[ ] KR_160607_SWATH_6600_CD1_WK10_1_Urea.wiff 2023-03-11 17:12 11M
[ ] KR_160607_SWATH_6600_CD1_WK10_1_Urea_profile.mzML 2023-03-11 14:05 5.6G
[TXT] KR_160607_SWATH_6600_CD1_WK10_1_Urea_profile_out.tsv 2023-03-11 11:59 279M
[ ] KR_160607_SWATH_6600_CD1_WK10_2_Urea.wiff 2023-03-11 16:03 11M
[ ] KR_160607_SWATH_6600_CD1_WK10_2_Urea_profile.mzML 2023-03-11 11:47 5.8G
[TXT] KR_160607_SWATH_6600_CD1_WK10_2_Urea_profile_out.tsv 2023-03-11 18:10 313M
[ ] KR_160607_SWATH_6600_CD1_WK10_3_Urea.wiff 2023-03-11 14:16 11M
[ ] KR_160607_SWATH_6600_CD1_WK10_3_Urea_profile.mzML 2023-03-11 13:11 5.4G
[TXT] KR_160607_SWATH_6600_CD1_WK10_3_Urea_profile_out.tsv 2023-03-11 16:48 296M
很多种不同数据格式的文件公开
一个样品可以有很多种不同数据格式的文件公开,比如:https://www.ebi.ac.uk/pride/archive/projects/PXD051600
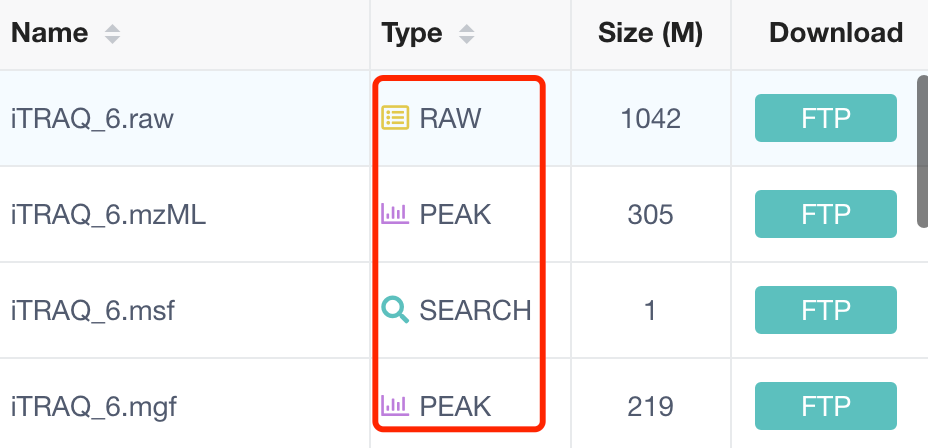
很多种不同数据格式的文件公开
在蛋白质组学研究中,质谱数据文件是分析蛋白质表达和鉴定的关键。基于您提供的文件列表,这些文件代表了不同的数据类型和分析阶段。以下是每种文件类型及其可能的区别:
- iTRAQ_6.raw:
- 这是原始的质谱数据文件,通常是直接从质谱仪获得的未经处理的数据。
.raw文件包含了实验中捕获的所有信息,包括离子的强度和质量。
- 这是原始的质谱数据文件,通常是直接从质谱仪获得的未经处理的数据。
- iTRAQ_6.mzML:
.mzML文件是一种用于存储质谱数据的标准化格式,它包含了从原始.raw文件中提取的信息,如肽段的质量和保留时间,但通常不包含原始的谱图。
- iTRAQ_6.msf:
.msf文件通常由Mascot搜索引擎生成,是二进制文件,包含了肽段指纹图谱数据,这些数据用于与数据库中的蛋白质序列进行比对。
- iTRAQ_6.mgf:
.mgf文件是Mascot生成的文件格式,通常包含了质谱数据的峰值列表,这些数据用于肽段鉴定和蛋白质序列匹配。
这些文件在蛋白质组学数据分析的流程中扮演着不同的角色,从原始数据的获取到肽段和蛋白质的鉴定,再到最终的生物信息学分析。每种文件格式都有其特定的用途,通常需要特定的软件工具来读取和分析。在处理这些数据时,研究人员需要根据实验设计和分析目标选择合适的文件和工具。
可以同时跑多个搜库软件
一个蛋白质组样品也可以跑多个搜库软件,比如: https://www.ebi.ac.uk/pride/archive/projects/PXD024174
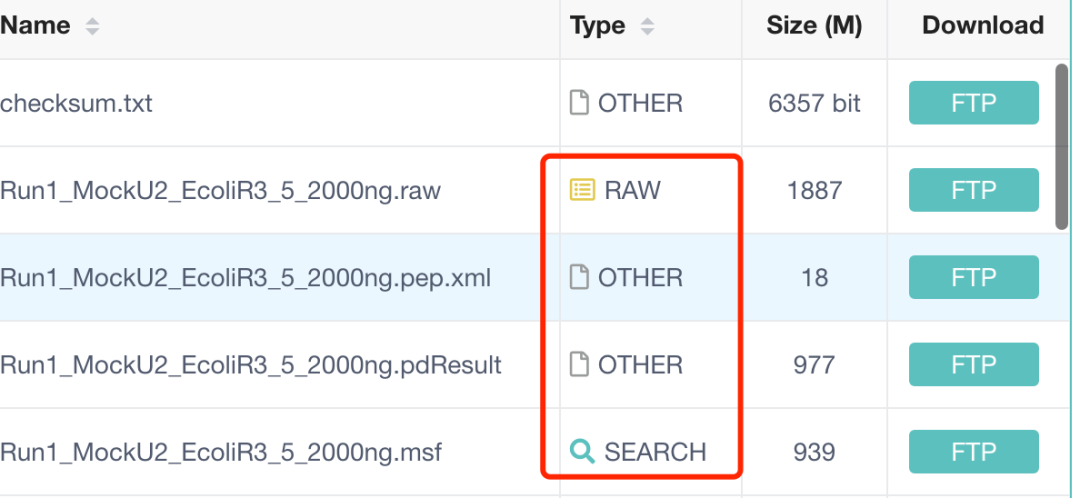
跑多个搜库软件
搜库的目的是为了将质谱数据与已知的蛋白质序列进行比对,从而鉴定样品中的蛋白质。搜索引擎(如Mascot、SEQUEST等)会使用这个数据库来分析质谱数据,尝试匹配观察到的肽段质量和序列与数据库中的蛋白质序列,以鉴定蛋白质。这些文件和数据库的创建是蛋白质组学数据分析的关键步骤,研究人员通过这些步骤可以识别和定量样品中的蛋白质,进而研究生物学问题或疾病机制。
- Run1_MockU2_EcoliR3_5_2000ng.raw:
- 这是一个原始的质谱数据文件,通常包含了质谱仪在实验中捕获的所有原始信号和信息。
- 文件名中的“2000ng”可能指的是样品中蛋白质的量,即2000纳克。
- Run1_MockU2_EcoliR3_5_2000ng.pep.xml:
.pep.xml文件通常包含了肽段鉴定信息,这些信息可能是由搜索引擎如Mascot生成的。- 这个文件可能包含了肽段的匹配结果、得分、覆盖的蛋白质序列等信息。
- Run1_MockU2_EcoliR3_5_2000ng.pdResult:
- 这个文件可能是由Proteome Discoverer软件生成的,包含了蛋白质鉴定的结果。
.pdResult文件通常包含了蛋白质鉴定的详细统计数据、鉴定的置信度、蛋白质列表等。
- Run1_MockU2_EcoliR3_5_2000ng.msf:
.msf文件是Mascot搜索引擎的输出文件,包含了肽段匹配的详细结果。- 这个文件可能包含了肽段的质量和序列、匹配的蛋白质、得分以及统计信息。
因为上面的这个案例是大肠杆菌,所以还需要附带该物种的参考基因组对应的蛋白质组的每个蛋白质的序列信息:
Mock_Comm_RefDB_V3_Clustered95.fasta
Mock_Comm_RefDB_V3.fasta
如果是人类和小鼠,基本上公开数据就太多太多了,可以省略物种的参考基因组对应的蛋白质组的每个蛋白质的序列信息。
再比如:https://www.ebi.ac.uk/pride/archive/projects/PXD051384
All data were quantified and searched against a Uniprot rat database (July 2014) using MaxQuant (v.1.5.3.28) as well as ProteomeDiscoverer/Mascot.
两个搜库软件就给出来了两个蛋白质表达量矩阵文件:
MS173180LUM_Teresina_Laragione_complete_analysis_PD.xlsx
MS173180LUM_Teresina_Laragione_complete_analysis_MQ.xlsx
需要理解蛋白质组学的上下游数据文件格式
以下是一些流行的蛋白质组学数据处理软件的概览,包括它们的开发者、大致开发年代、输入数据类型、输出文件格式等信息:
- ProteomeDiscoverer (PD)
*.xlsx:用于肽段和蛋白质的导出,通常包含鉴定和定量信息- 其他格式:PD支持多种输出格式,用于进一步分析或提交至数据库
- 开发者:Thermo Fisher Scientific
- 年代:2010年代初发布
- 输入数据:质谱原始数据文件,如Thermo的.raw格式
- 输出文件格式:
- 特点:提供全面的数据分析和可视化工具,需要布局配置以优化搜索和结果展示。
- MaxQuant
evidence.txt:包含肽段鉴定的证据proteinGroups.txt:包含蛋白质组的鉴定信息- 开发者:MPI of Biochemistry in Martinsried, Germany
- 年代:2006年首次发布
- 输入数据:质谱原始数据文件,支持多种格式如.mzML和.mzXML
- 输出文件格式:
- 特点:集成了搜索、鉴定、定量和标签自由量化等功能,用户友好,支持大量数据集的处理。
- FragPipe
combined_peptide.tsv:合并的肽段报告combined_protein.tsv:合并的蛋白质报告- 开发者:Broad Institute
- 年代:2016年发布
- 输入数据:质谱原始数据文件
- 输出文件格式:
- 特点:专为高通量数据分析设计,支持自动化处理和并行计算。
- Skyline
*.csv:肽段报告,通常与MSstats软件结合使用- 开发者:University of Washington
- 年代:2005年左右开发
- 输入数据:质谱数据,需要MSstats安装
- 输出文件格式:
- 特点:主要用于靶向蛋白质组学,特别是SRM(选择反应监测)实验。
- DIA-NN/DIA-NN-LFQ
*.tsv:肽段报告- 开发者:University of Michigan
- 年代:2015年左右开发
- 输入数据:质谱原始数据文件,支持DIA(数据独立采集)模式
- 输出文件格式:
- 特点:专为DIA数据分析设计,提供标签自由和标签依赖的定量分析。
- mzTab
*.mzTab:一种标准化的表格格式,用于存储蛋白质组学数据- 开发者:Proteomics Standards Initiative (PSI)
- 年代:2010年代中期提出
- 输入数据:质谱数据和搜索结果
- 输出文件格式:
- 特点:mzTab旨在促进数据的共享和再利用,不跟踪多肽边界(MBR)。
请注意,上述信息可能随时间和软件更新而变化。每种软件都有其特定的应用场景和优势,研究人员通常会根据实验设计、数据类型和个人偏好选择合适的工具。此外,一些软件可能需要与其他工具或数据库配合使用,以完成特定的分析流程。
太多蛋白质搜库软件
简单的整理了一下蛋白质组搜库软件列表,以及它们的网页链接:
- MSFragger:
- 描述:片段离子索引方法,显著提高搜库速度,全面识别肽及其所有修饰形式。
- 链接:MSFragger
- MyriMatch:
- 描述:基于多元超几何分布的统计模型,强调匹配强烈峰值,提高辨别能力。
- 链接:MyriMatch
- Census:
- 描述:与多种质谱数据兼容,使用强大算法解决低质量测量值,支持多种输入文件格式。
- 链接:Census
- MZmine 2:
- 描述:用于处理、可视化和分析质谱数据,特别强调易用性和高分辨率质谱数据处理。
- 链接:MZmine 2
- MaxQuant:
- 描述:广泛应用的质谱数据分析平台,支持label free、MS1-level labeling和同位素标记技术。
- 链接:MaxQuant
- OMSSA:
- 描述:高效、灵敏、特异的质谱搜索算法,用于肽段鉴定。
- 链接:OMSSA
- OpenMS:
- 描述:质谱分析的软件框架,支持从基本数据结构到复杂数据分析算法。
- 链接:OpenMS
- Comet:
- 描述:开源MS/MS序列数据库搜索工具,基于广泛使用的原始搜索工具。
- 链接:Comet
- SuperHirn:
- 描述:用于处理高分辨率LC-MS数据,提供特征提取、定量、相似性分析等功能。
- 链接:SuperHirn
- MS-GF+:
- 描述:敏感且通用的数据库搜索工具,适用于多种类型的质谱数据和仪器配置。
- 链接:MS-GF+
- InsPecT:
- 描述:从串联质谱数据中识别翻译后修饰,使用数据库过滤器和新算法评分和验证修饰。
- 链接:InsPecT
- SpectraST:
- 描述:利用谱图库进行肽段鉴定,集成到Trans Proteomic Pipeline中,提供概率分配、定量和数据可视化功能。
- 链接:SpectraST
- MS Amanda:
- 描述:适合高精度质谱数据的评分算法,提供更多谱图解释,免费获取。
- 链接:MS Amanda
- Protein Pilot:
- 描述:使用Paragon算法,提供新型搜库引擎,用户界面友好,有效搜索空间大。
- 链接:Protein Pilot
- msInspect:
- 描述:开源应用程序,用于分析多个LC-MS实验测量值,支持label free和同位素标记方法定量。
- 链接:msInspect
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-06-02,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号