一键完成三种差异分析:DEseq2, edgeR and limma

一键完成三种差异分析:DEseq2, edgeR and limma

生信菜鸟团
发布于 2024-06-11 17:38:50
发布于 2024-06-11 17:38:50
limma、edgeR、DESeq2原理
Limma基于线性模型,通过使用贝叶斯方法估计每个基因的差异方差。它使用经验贝叶斯方法来将信息从所有基因中借用,特别是在样本较少时提高估计的稳定性。
edgeR基于负二项分布模型。它使用贝叶斯方法通过适应组内变异估计提高估计的稳定性。edgeR考虑了基因的丰度和变异性,使其更适用于RNA-Seq数据。
DESeq2基于负二项分布的模型。它通过使用贝叶斯方法来考虑样本间的差异以及基因表达的离散性。
1. 安装和加载所需的包
.libPaths(
c(
'/home/rootyll/seurat_v5/',
"/usr/local/lib/R/site-library",
"/usr/lib/R/site-library",
"/usr/lib/R/library"
)
)
# 安装必要的包,如果尚未安装
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
#BiocManager::install(c("Biobase", "limma", "edgeR", "DESeq2", "biomaRt", "VennDiagram"))
如果安装失败,需要:
wget -c https://codeload.github.com/montilab/BS831/zip/refs/heads/master
unzip master
devtools::install_local("~/gzh/20240608_limma_deseq2_edegr/BS831-master/",dependencies = TRUE)
# 加载必要的包
#devtools::install_github("montilab/BS831",dependencies = TRUE)
library(BS831)
library(Biobase)
library(limma)
library(edgeR)
library(DESeq2)
library(biomaRt)
library(VennDiagram)
2. 定义包的封装函数
run_deseq <- function(eset, class_id, control, treatment) {
control_inds <- which(pData(eset)[, class_id] == control)
treatment_inds <- which(pData(eset)[, class_id] == treatment)
eset.compare <- eset[, c(control_inds, treatment_inds)]
colData <- data.frame(condition=as.character(pData(eset.compare)[, class_id]))
dds <- DESeqDataSetFromMatrix(exprs(eset.compare), colData, formula( ~ condition))
dds$condition <- factor(dds$condition, levels=c(control,treatment))
dds_res <- DESeq(dds)
res <- results(dds_res)
res$dispersion <- dispersions(dds_res)
return(res)
}
run_edgeR <- function(eset, class_id, control, treatment) {
control_inds <- which(pData(eset)[, class_id] == control)
treatment_inds <- which(pData(eset)[, class_id] == treatment)
eset.compare <- eset[, c(control_inds, treatment_inds)]
condition <- as.character(pData(eset.compare)[, class_id])
y <- DGEList(counts=exprs(eset.compare), group = condition)
y <- calcNormFactors(y)
y <- estimateGLMCommonDisp(y)
y <- estimateGLMTrendedDisp(y)
y <- estimateGLMTagwiseDisp(y)
et <- exactTest(y)
res <- topTags(et, n = nrow(eset.compare), sort.by = "none")
return(res)
}
run_limma <- function(eset, class_id, control, treatment) {
control_inds <- which(pData(eset)[, class_id] == control)
treatment_inds <- which(pData(eset)[, class_id] == treatment)
eset.compare <- eset[, c(control_inds, treatment_inds)]
condition <- as.character(pData(eset.compare)[, class_id])
colData <- data.frame(condition=as.character(pData(eset.compare)[, class_id]))
design <- model.matrix(~ 0 + factor(condition))
colnames(design) <- levels(factor(condition))
fit <- lmFit(eset.compare, design)
command_str <- paste("makeContrasts(",
"(", treatment , "-", control, ")",
",levels = design)", sep = "")
contrast.matrix <- eval(parse(text=command_str))
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
res <- topTable(fit2, coef=1, adjust="BH", sort.by = "none", number=Inf)
return(res)
}
3. 加载数据集
如果是你自己的数据,需要整理成这样的数据格式
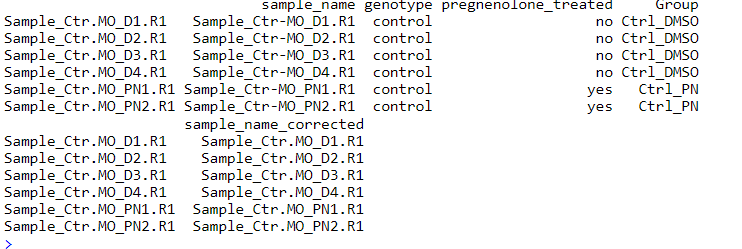
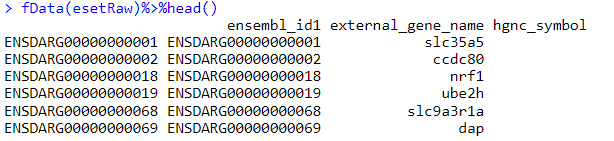
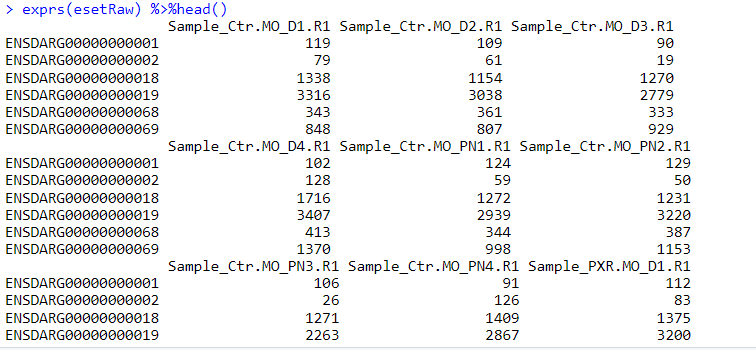
4. 运行
#4运行----
## see wrappers at code/diffanalWrappers.R
## run deseq2
res_deseq2 <- run_deseq(eset=esetRaw , class_id="Group", control="Ctrl_DMSO", treatment="Ctrl_PN")
## run edgeR htseq counts
res_edgeR <- run_edgeR(eset=esetRaw , class_id="Group", control="Ctrl_DMSO", treatment="Ctrl_PN")
## run limma with cufflinks fpkm log2-transformed data
res_limma <- run_limma(eset=esetFPKM , class_id="Group", control="Ctrl_DMSO", treatment="Ctrl_PN")
写在最后
为了更好的理解三个不同的r包,还是希望读者可以亲自使用不同的r包,去体验一下整个流程。在已经熟悉r包的基础上,再使用函数随时调用
不足之处,欢迎指正——生信小博士
参考:
https://bioconductor.org/packages/release/workflows/vignettes/RNAseq123/inst/doc/limmaWorkflow.html
https://montilab.github.io/BS831/articles/docs/DiffanalysisRNAseqComparison.html
https://montilab.github.io/BS831/articles/docs/Diffanalysis.html本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-06-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号