课前准备---单细胞数据检测SNV(变异、插入、缺失、等位基因连锁)
原创
课前准备---单细胞数据检测SNV(变异、插入、缺失、等位基因连锁)
原创
追风少年i
修改于 2024-09-06 21:39:21
修改于 2024-09-06 21:39:21
作者,Evil Genius
单细胞检测变异的分析已经分享了很多,全部发的高分文章。
日前外显子的课程已经安排上了,但是不一定能上,可能外显子分析对大家来讲不太重要吧,但更可能是大家都会分析,不管上不上,先好好备课吧(正好也偷个懒)。
不过提醒大家一句,正因为做的人少,才是机会,表达信息的分析已经烂大街了,大家是时候面向新的方向研究了。
今日话题:单细胞数据检测SNV
单细胞RNA测序(scRNA-seq)在单细胞分辨率上提供了对转录组的独特分析,揭示了巨大的细胞异质性。尽管这种类型的数据包含了细胞转录组的丰富信息,但大多数研究只关注基因表达,而没有处理其他重要方面,如单核苷酸变异(SNV)或等位基因特异性表达。
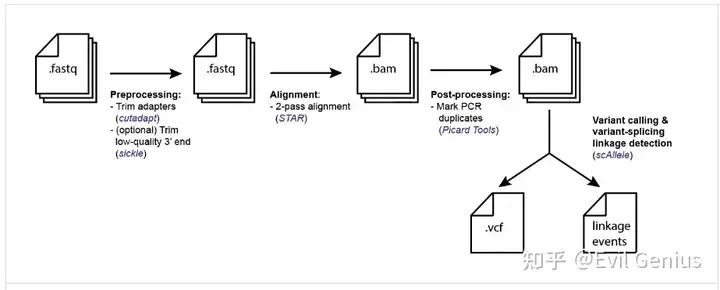
检测框架
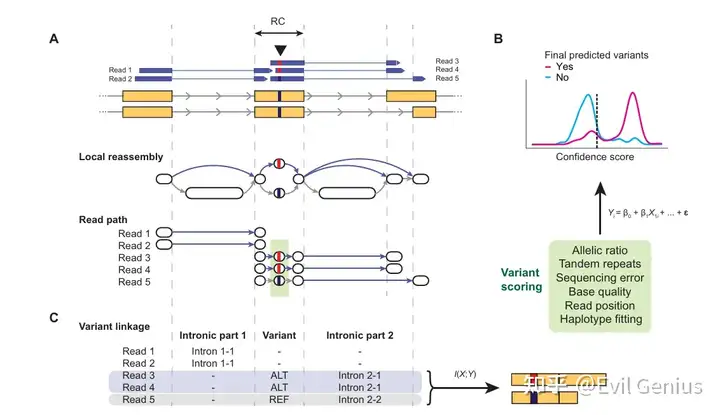
分析示例,细胞系的检测可靠性
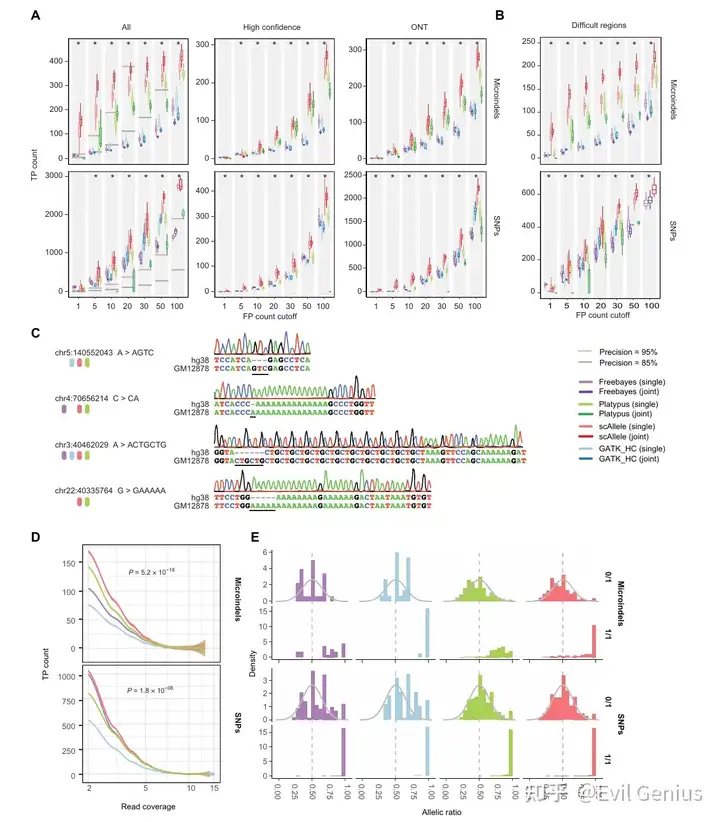
分析示例,检测变异之间的联动性,即等位基因连锁分析
尽管单个细胞的测序深度有限,但典型的scRNA-seq数据集包含大量细胞。因此,合并来自多个细胞的数据可以有效地增加可测试事件的数量,用于遗传变异和剪接之间的连锁分析。
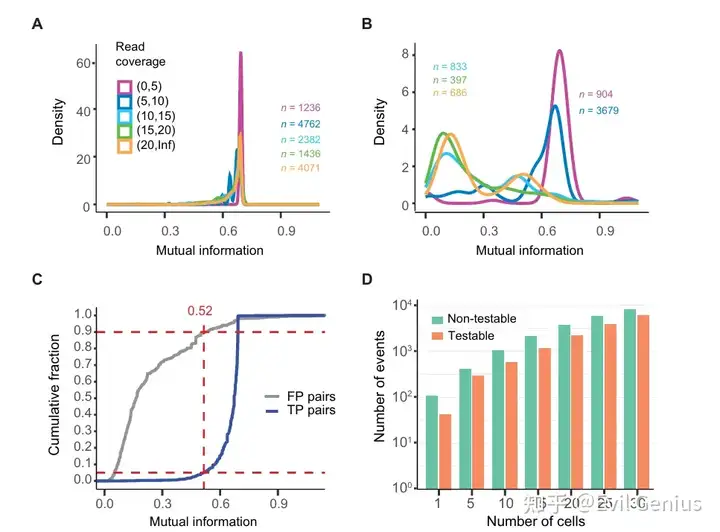
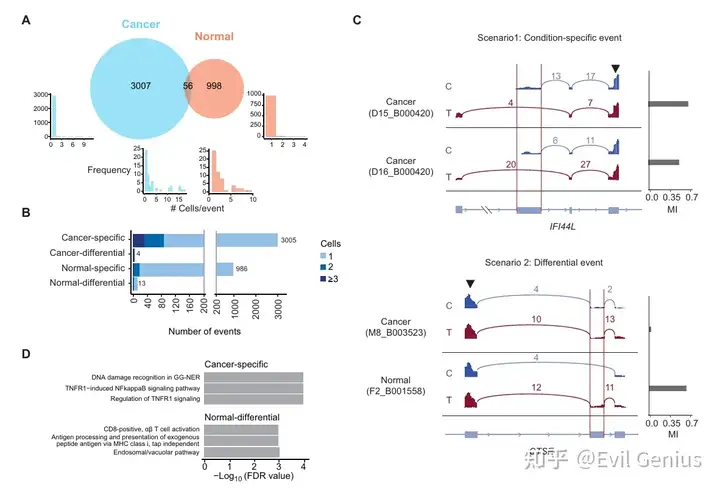
分析示例:单细胞SNV检测揭示了肺癌细胞中核苷酸变异和等位基因特异性剪接事件
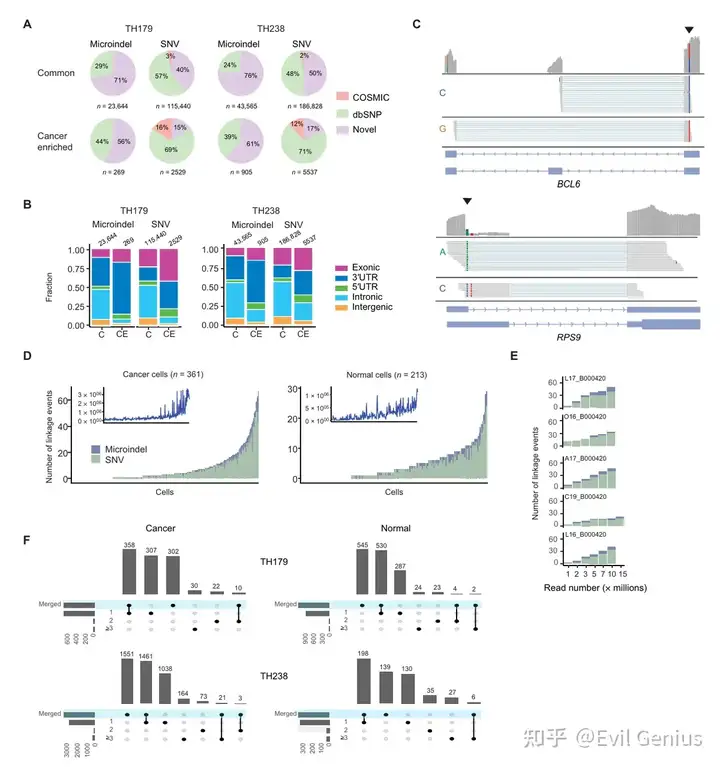
分析示例:癌症和正常细胞表现出独特和不同的等位基因特异性剪接事件
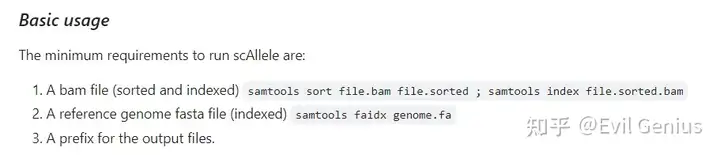
代码示例
scAllele -b file.sorted.bam -g genome.fa -o path/to/output_prefix Filtering variants
scAllele
-b testdata/gm12878.chr21.bam
-g testdata/hg38.chr21.fa
-o path/to/output_prefix
--AC=3
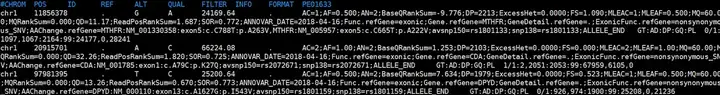
--DP=5输出的文件
下游分析

生活很好,有你更好
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号