Benchmarking:新鲜出炉的单细胞自动化癌细胞注释


今天给大家介绍一款新鲜出炉的基准测试类文章,于2025年1月发表在预印本biorxiv上,标题为:《Benchmarking of automated cancer cell annotation methods for scRNA-seq data reveals Consensus annotation as the preferred method》。

这篇文献的核心内容是关于 scRNA-seq 数据中癌细胞注释方法的基准测试研究。研究比较了基于参考数据(Reference-based)和基于拷贝数变异(CNV-based)的两种主要癌细胞注释方法,并提出了一种新的共识注释(Consensus annotation)方法,该方法结合了前两种方法的优势,能够更准确地识别肿瘤细胞。
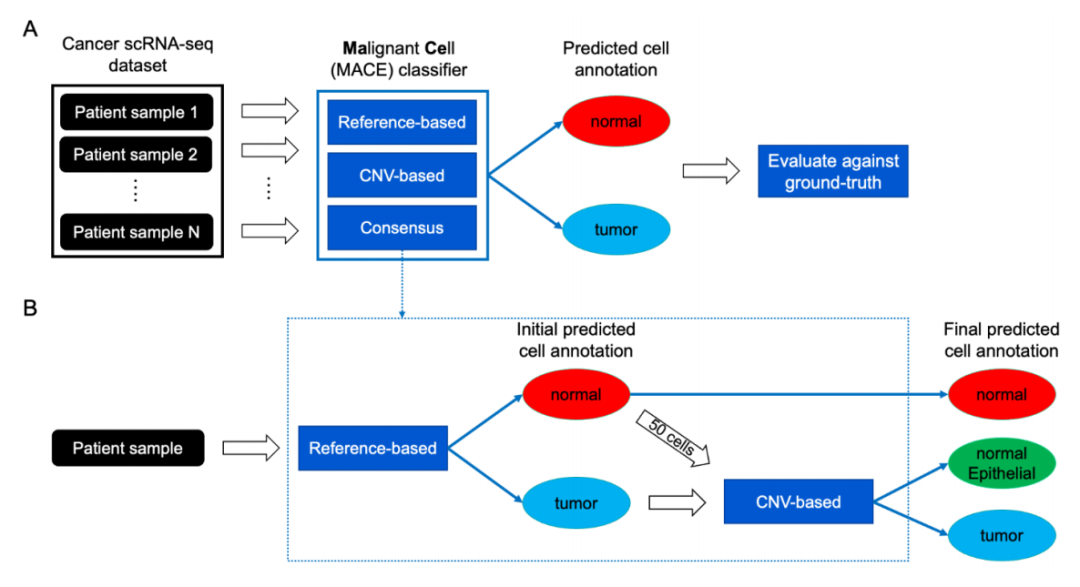
Benchmarking pipeline
现有方法的局限性
传统的手动注释方法依赖于先验知识和标记基因,存在主观性和不完整性。自动化的分类器方法虽然更一致和可扩展,但在癌症研究中区分复杂的肿瘤微环境方面存在挑战。
测试数据集
研究使用了20个癌症scRNA-seq数据集,涵盖9种癌症类型,共379个样本
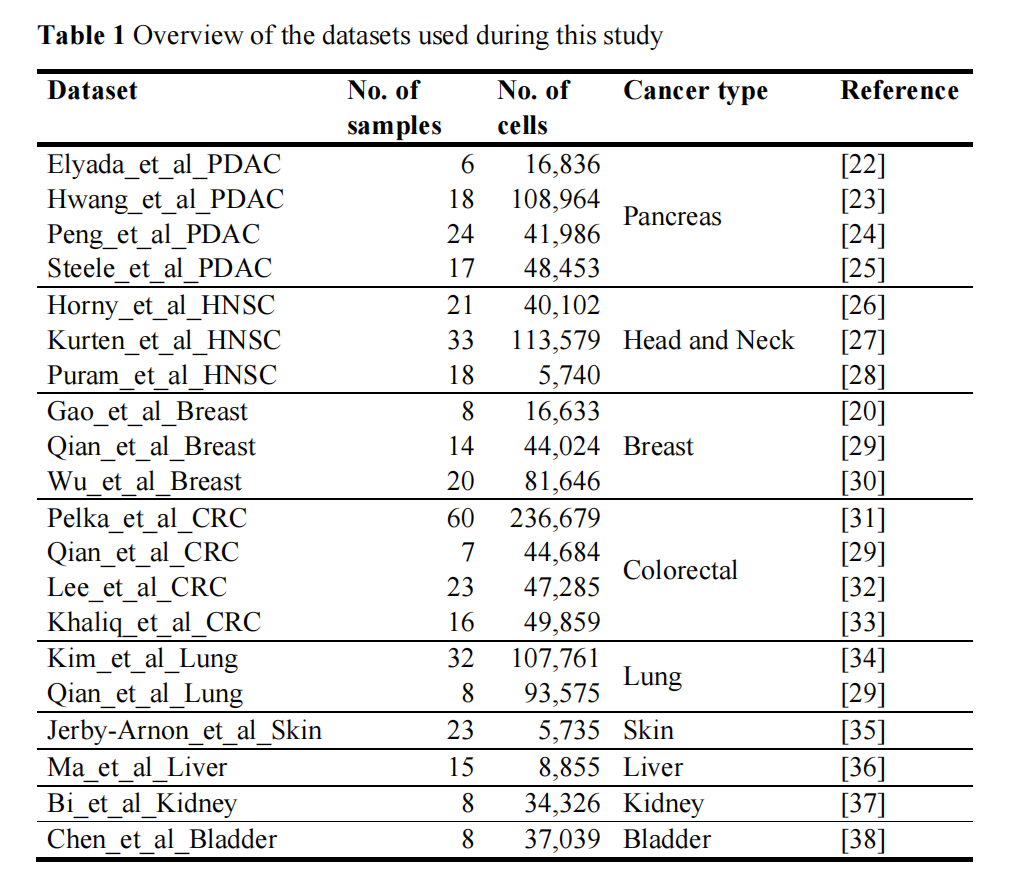
评估指标
主要使用精确度(Precision)作为评估指标,即正确注释为肿瘤细胞的真正肿瘤细胞与被错误注释为肿瘤细胞的正常细胞的比例。
- 基于参考数据的方法(Reference-based):使用scATOMIC,通过已标注的癌症数据训练分类器来注释新数据集。https://github.com/abelson-lab/scATOMIC
- 基于拷贝数变异的方法(CNV-based):使用SCEVAN,通过基因表达数据推断拷贝数变异来识别肿瘤细胞。
- 共识注释方法(Consensus annotation):结合上述两种方法,只有当两种方法都同意某个细胞为肿瘤细胞时,该细胞才被注释为肿瘤细胞。

基于参考数据的方法与基于拷贝数变异(CNV)的方法的基准测试结果
散点图展示了在9种癌症类型中,基于参考数据的方法和基于CNV的方法的精确度评分。图中的每个点代表一个患者样本,位于上三角区域的点越多,表示基于参考数据的方法性能越高;而位于下三角区域的点越多,则表示基于CNV的方法性能越高。
性能差异:不同癌症类型中,两种主要方法的性能表现不同。例如,在头颈癌、乳腺癌、结直肠癌、肺癌和肾癌中,基于参考数据的方法表现更好;而在胰腺癌和肝癌中,基于拷贝数变异的方法表现更好。
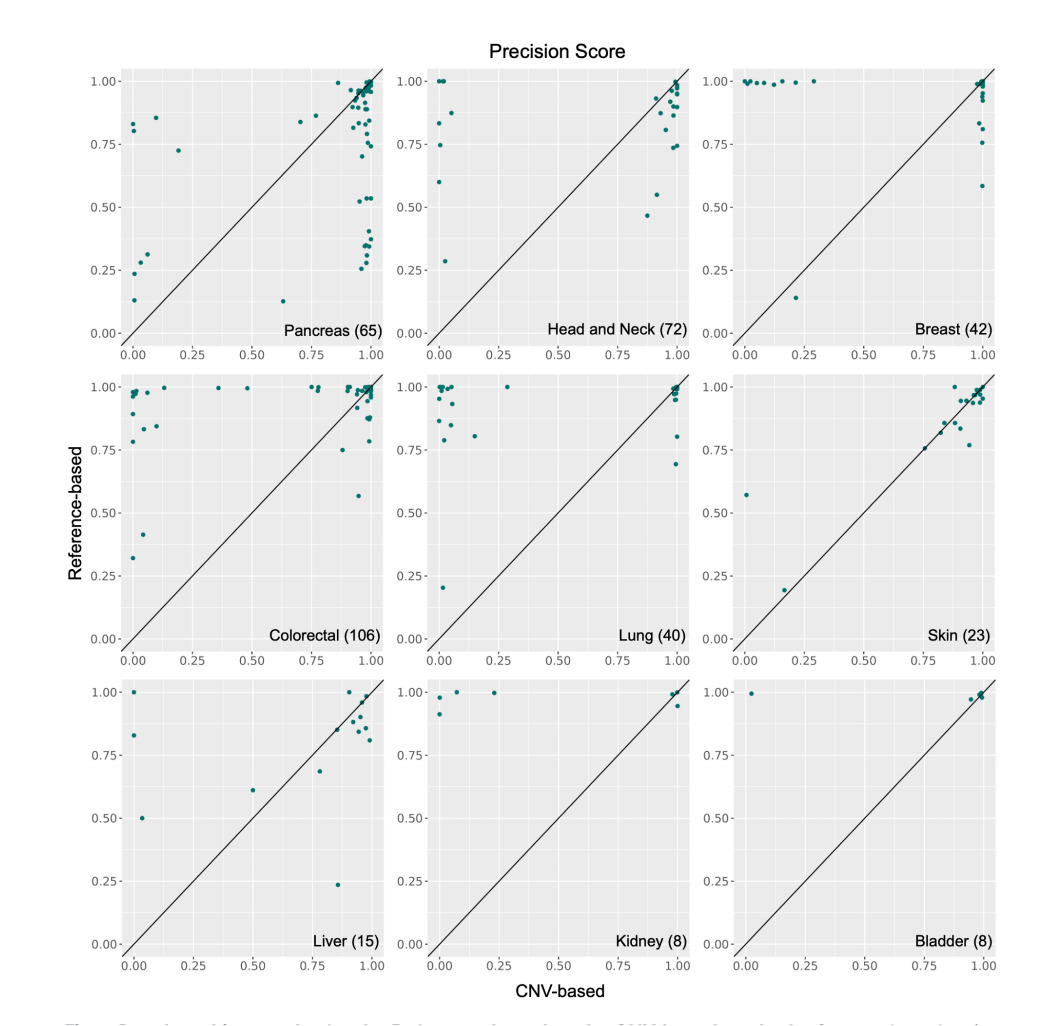
Fig.2
共识注释方法结合了其他方法的优势
- (A) Peng_et_al_PDAC 数据集的UMAP可视化,根据作者的细胞类型注释进行着色,并与基于参考数据、基于CNV和共识方法产生的MACE注释进行比较。
- (B) Qian_et_al_Breast 数据集的UMAP可视化,同样根据作者的细胞类型注释进行着色,并与上述三种方法产生的MACE注释进行比较。
- (C) 箱线图比较了所有方法的性能,基于精确度评分,表明共识方法在9种不同癌症类型中产生了最精确的注释。
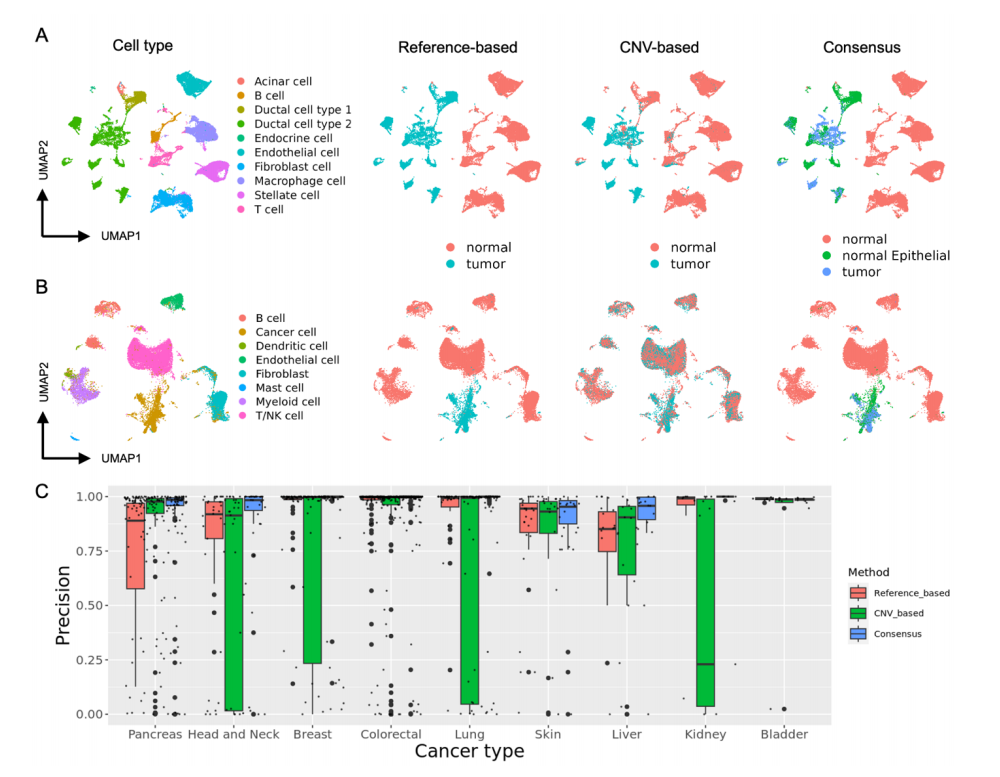
共识注释的优势
共识注释方法在所有癌症类型中都表现出了最高的精确度,能够有效减少假阳性注释,同时准确区分正常上皮细胞和恶性上皮细胞。
CNV分析验证:共识注释方法识别的肿瘤细胞具有明确的CNV特征,与正常上皮细胞的二倍体CNV特征形成对比。这进一步证明了共识注释方法的有效性。
该代码可在GitLab上的MACE R包中获取:https://gitlab.com/genmab-public/mace/
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-02-01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号