数据集“变脸术”?曾老板给的双胞胎数据集之谜


曾老板给我发了两个有意思的数据集,这两个数据集具有不同的GSE编号,联系单位人等什么的都不一样,但是他们的样本总数,以及样本构成实在是太相似了,不得不让人怀疑是一个数据集重新上牌号后摇身一变,变成了一个新的数据集。看我们如何用生物信息学的手段来侦查它!
这两个数据集分别是:2012年update 在GEO上的 GSE41258,以及 2015年 update的GSE68468
- https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41258
- https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68468
GSE41258数据
当然,首先看先发表的那个,GSE41258 这个数据集总共有390个样本,主要有结肠癌等相关样本,关联了两个paper,通讯联系人为 Michal Sheffer:
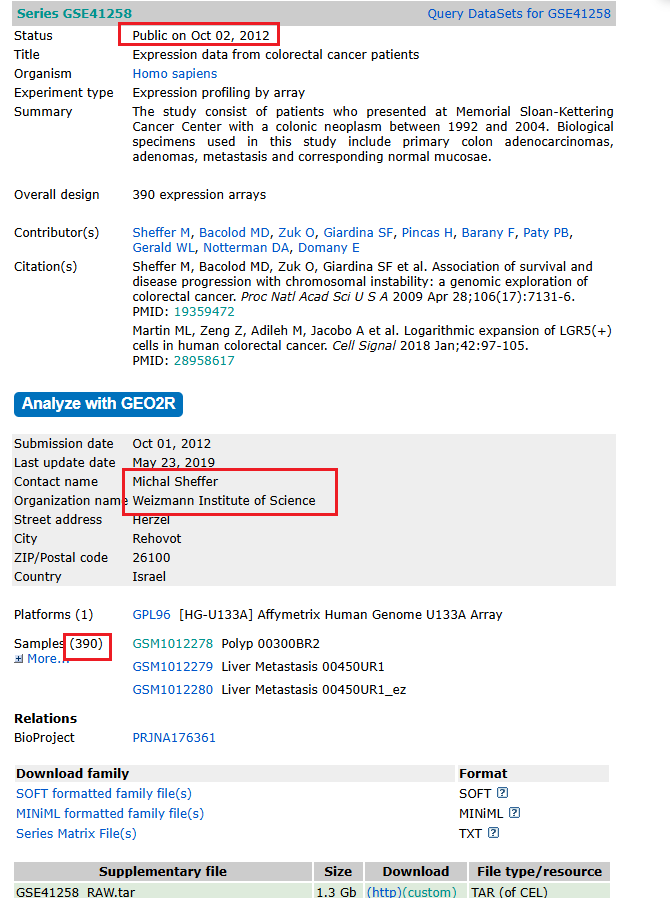
简单探索一下这个数据,看下表型:
## 加载R包
library(AnnoProbe)
library(GEOquery)
library(ggplot2)
library(ggstatsplot)
library(patchwork)
library(reshape2)
library(stringr)
library(limma)
library(tidyverse)
getOption('timeout')
options(timeout=10000)
options(scipen = 1000)
## 1.获取并且检查表达量矩阵
## ~~~gse编号需修改~~~
gse_number <- "GSE41258"
dir.create(gse_number)
setwd(gse_number)
getwd()
list.files()
#gset <- geoChina(gse_number)
gset <- getGEO(gse_number, destdir = '.', getGPL = T)
gset[[1]]
a <- gset[[1]]
## 2.样本分组
## 挑选一些感兴趣的临床表型。
pd <- pData(a)
pd$title
pd$`anatomic location:ch1`
table(pd$`t:ch1`)
pd$`tissue:ch1`
as.data.frame(table(pd$`tissue:ch1`))
## ~~~分组信息编号需修改~~~
group_list <- sub("^(.*\\s).*", "\\1", pd$title) # 提取最后一个空格之前的内容
group_list <- trimws(group_list, "right")
group_list
as.data.frame(table(group_list))
每种类型的样本数如下:
as.data.frame(table(group_list))
group_list Freq
1 Cell line 12
2 Liver Metastasis 47
3 Lung Metastasis 20
4 Microadenoma 2
5 Normal Colon 54
6 Normal Liver 13
7 Normal Lung 7
8 Polyp 48
9 Polyp, high grade 1
10 Primary Tumor 186
GSE68468数据
GSE68468 这个数据集也有390个样本,也是结肠癌等相关样本,关联了7个paper,通讯联系人为 Mervi Heiskanen:
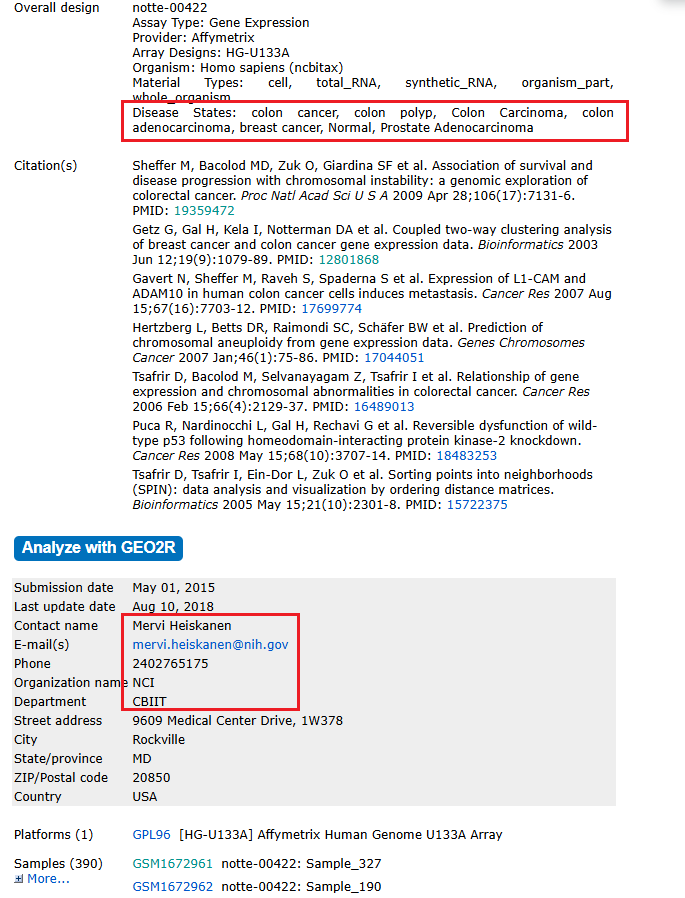
简单探索一下:
## 1.获取并且检查表达量矩阵
## ~~~gse编号需修改~~~
gse_number <- "GSE68468"
dir.create(gse_number)
setwd(gse_number)
getwd()
list.files()
#gset <- geoChina(gse_number)
gset <- getGEO(gse_number, destdir = '.', getGPL = T)
gset[[1]]
a <- gset[[1]]
## 2.样本分组
## 挑选一些感兴趣的临床表型。
pd <- pData(a)
colnames(pd)
pd$title
pd$`cell_line:ch1`
table(pd$`disease_location:ch1`)
table(pd$`disease_state:ch1`)
pd$`histology:ch1`
as.data.frame(table(pd$`histology:ch1`))
每种类型的样本数如下:
as.data.frame(table(pd$`histology:ch1`))
Var1 Freq
1 breast cancer 1
2 colon adenocarcinoma 2
3 colon cancer 186
4 Colon Carcinoma 7
5 colon carcinoma metastatic to the liver 47
6 colon carcinoma metastatic to the lung 20
7 colon polyp 48
8 high grade dysplasia 1
9 microadenoma 2
10 normal colon mucosa 55
11 normal liver 13
12 normal lung 7
13 Prostate Adenocarcinoma 1
与上面可以说几乎是一样的,这也太巧了吧!
用生物信息学手段来侦查
要比较两个数据集中的样本是不是一样的,我们可以将两个数据集的样本表达矩阵合并在一起,然后计算样本间的相关性,通过相关性基本上就能判断了。
首先,这两个数据集都提供了芯片原始数据CEL文件,平台都是GPL96,为了保持数据的一致性,我们这里选择对CEL进行同样的RMA标准化。
下载下面这两个文件,使用RMA标准化得到表达矩阵:
GSE68468_RAW.tar
GSE41258_RAW.tar
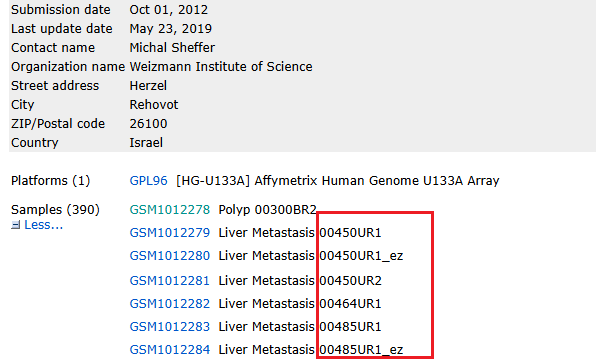
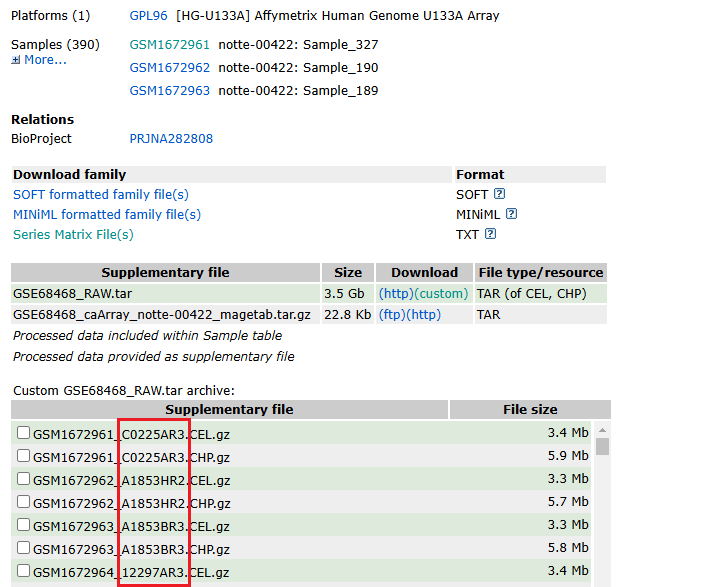
新线索:我发现这两个数据具有同一个 奇怪的编号:
GSE41258的:
GSE68468的:
那就利用这个ID号将两个数据联合起来看看:
读取两次的表达矩阵:
## 2012年上传的 GSE41258
load("../../2012-GSE41258-390-ColonCancer/GSE41258/step1_output_rma.Rdata")
ls()
dat[1:5, 1:5]
dim(dat)
colnames(pd)
# 提取最后一个空格之后的内容
pd$id <- sub("^.*\\s(.*)", "\\1", pd$title)
pd$group <- sub("^(.*\\s).*", "\\1", pd$title)
pd$group <- trimws(pd$group, "right")
phe1 <- pd[, c("geo_accession", "id", "group")]
phe1
datGSE41258 <- dat
datGSE41258[1:5, 1:5]
## 2015年上传的 GSE68468
load("step1_output_rma.Rdata")
ls()
dat[1:5, 1:5]
datGSE68468 <- dat
datGSE68468[1:5, 1:5]
pd$`histology:ch1`
phe2 <- cbind(pheo, pd[pheo[,1], "histology:ch1"])
colnames(phe2) <- c("geo_accession", "id", "group")
head(phe2)
简单合并:
## 合并
id <- intersect(phe1[,2], phe2[,2])
length(id)
merge <- merge(phe1, phe2,by="id", suffixes = c(".GSE41258", ".GSE68468") )
head(merge)
table(merge$group.GSE41258, merge$group.GSE68468)
merge$d1 <- paste0(merge$id, "-",merge$geo_accession.GSE41258)
merge$d2 <- paste0(merge$id, "-",merge$geo_accession.GSE68468)
## 绘制两个数据集之间的相关性
identical(rownames(datGSE41258), rownames(datGSE68468))
colnames(datGSE41258) <- merge[ match(colnames(datGSE41258), merge$geo_accession.GSE41258), "d1"]
colnames(datGSE68468) <- merge[ match(colnames(datGSE68468), merge$geo_accession.GSE68468), "d2"]
cor_dat <- cbind(datGSE41258, datGSE68468)
colnames(cor_dat)
grep("00300BR2",colnames(cor_dat),value = T)
grep("09518BR3",colnames(cor_dat),value = T)
dim(cor_dat)
cor_dat[1:10, 1:10]
计算相关性:
cor <- cor(cor_dat)
anno <- data.frame(gse=rep(c("GSE41258","GSE68468"), times=c(ncol(datGSE41258), ncol(datGSE68468) ) ) )
rownames(anno) <- colnames(cor_dat)
head(anno)
cor的结果:可以看到一些端倪了!
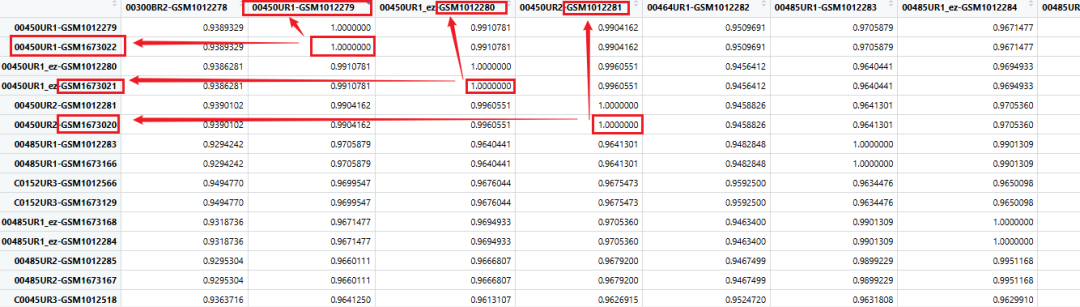
780个样本的矩阵太大,这里简单挑选20个样本绘图看下:
plotid <- merge[1:20, "id"]
plotid
cor_sub <- cor[, str_split(colnames(cor),"-",simplify = T)[,1] %in% plotid ]
cor_sub <- cor_sub[ str_split(rownames(cor),"-",simplify = T)[,1] %in% plotid, ]
p <- pheatmap(cor_sub, annotation_col = anno, display_numbers = T)
library(ggplotify)
p <- as.ggplot(p)
p
结果如下:
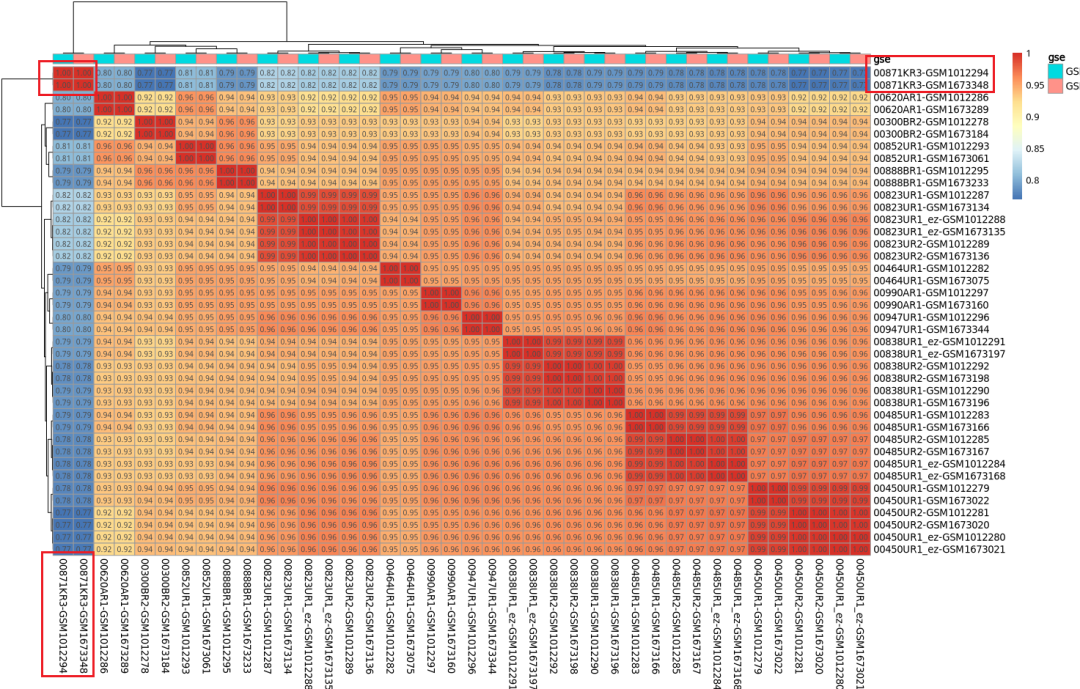
根据这张图,我们基本上已经可以下结论,GSE41258 和 GSE68468就是同一个数据集!
那为什么 GSE41258 要摇身一变,变成了 GSE68468呢?这背后有什么可疑的故事吗?
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-02-23,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号