单细胞转录组分析的10大软件/流程(上)

单细胞转录组分析的10大软件/流程(上)

用户11203141
发布于 2025-03-06 11:23:43
发布于 2025-03-06 11:23:43
面对如此多样化的工具,我们该如何筛选呢?别担心,我们有一个绝佳的参考资源网站:https://www.scrna-tools.org/。这个开源数据库收集了几乎所有的单细胞数据分析工具,让我们能够根据工具的引用次数来判断其影响力和实用性。
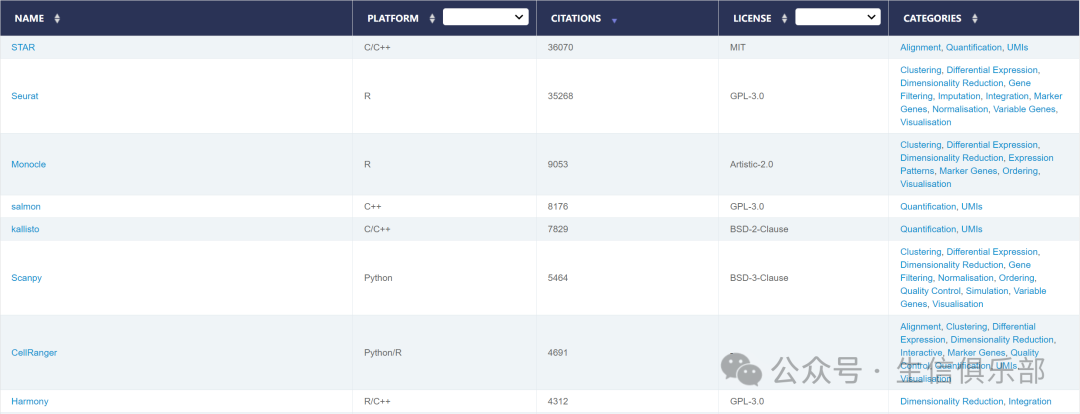
接下来,让我们聚焦于引用量最高的TOP10个工具,它们代表了当前单细胞数据分析领域的最佳实践:
Top1.STAR
STAR (Spliced Transcripts Alignment to a Reference)是一个用于RNA-seq数据的比对工具,其核心在于两阶段映射策略。在第一阶段,STAR会将RNA-seq读段与参考基因组比对,生成初步的比对结果和剪接点信息。在第二阶段,STAR会使用这些信息重新生成基因组索引,然后再进行一次更为精准的比对。这种两阶段映射不仅提高了比对的准确性,还能够发现新剪接点
亮点功能:
- 高速处理能力:STAR在处理大规模数据集时表现出色,能够在短时间内完成比对任务。相比其他同类工具,STAR的速度提升了50倍以上
- 高准确性:STAR能够精准识别RNA-seq读段中的剪接事件、融合基因和复杂的基因结构,适用于研究基因表达的动态变化和复杂的基因调控网络
- 灵活的用户自定义选项:STAR提供了多种参数供用户调整,如多线程处理、输出格式选择等,适应不同的研究需求
code:https://github.com/ConesaLab/acorde
适用场景: STAR特别适合需要高通量数据处理的科研项目,例如基因表达谱分析、转录本组装和变异检测。它的高速和高准确性使得研究者能够在短时间内得到可靠的数据,为后续的生物信息分析提供坚实基础。
2.Seurat
Seurat是单细胞RNA-seq数据分析的事实标准,自2015年推出以来,凭借其卓越的细胞亚型识别和基因表达模式分析能力,成为单细胞研究领域的领军工具。

工具简介: Seurat是一款用于处理和分析单细胞RNA测序数据的R包。其主要功能包括数据标准化、降维分析、聚类分析以及数据可视化。Seurat通过整合多种算法,使研究者能够从单细胞数据中提取有价值的信息,如识别细胞亚型、探索基因表达模式以及研究细胞间的异质性。
亮点功能:
- 多样的降维技术:Seurat提供了多种降维技术,如PCA、t-SNE和UMAP,帮助研究者在高维单细胞数据中识别关键特征 (academic.oup)。
- 强大的聚类分析能力:Seurat能够有效地将细胞分为不同的亚型,揭示细胞间的异质性,这对于研究细胞分化、肿瘤异质性等具有重要意义 (Springer)。
- 灵活的数据整合:Seurat支持不同数据集的整合分析,使得研究者可以结合多种数据源进行更全面的分析 (RS Blog)。
code:https://github.com/satijalab/seurat?tab=readme-ov-file
适用场景: Seurat被广泛应用于单细胞研究领域,适合研究者用于探索细胞异质性、识别新型细胞亚型,以及揭示基因表达的复杂模式。它为理解生物过程中的细胞行为提供了强有力的工具。
Top3:Monocle
Monocle于2014年推出,是一款专注于单细胞转录组数据分析的工具,特别是用于研究细胞发育轨迹和基因表达动态变化。它帮助研究人员揭示不同细胞类型在发育过程中的转变,捕捉细胞异质性。
工具简介: Monocle是一款开源R包,旨在通过单细胞RNA测序数据分析细胞的发育轨迹和基因表达模式。它通过“伪时间”(pseudotime)分析,帮助研究者理解细胞在发育过程中的进程。Monocle还提供了识别差异表达基因的功能,有助于揭示在特定发育阶段或细胞类型中起关键作用的基因。
亮点功能:
- 伪时间分析:Monocle能够通过伪时间分析方法,对单细胞数据进行排序,推断出细胞从一种状态到另一种状态的转变过程,帮助研究者理解细胞分化和发育的动态过程。
- 基因表达动态分析:Monocle可以识别出在不同发育阶段或细胞状态下表达水平变化显著的基因,为研究细胞分化中的关键调控因子提供了重要线索。
- 灵活的输入数据:Monocle能够处理多种形式的单细胞RNA测序数据,并支持不同数据集之间的整合分析 (GitHub)
Code: https://github.com/cole-trapnell-lab/monocle-release
适用场景: Monocle广泛应用于研究发育生物学、再生医学和肿瘤学领域,特别适合于探索细胞命运决定和发育过程中的基因表达变化。
TOP4:salmon
salmon是2017年推出的一款基因表达定量工具,采用创新的概率模型,为高通量测序数据的快速而准确的分析提供了解决方案。
工具简介: salmon是一款独立的软件包,专门用于RNA-seq数据的转录本定量。它使用了基于序列概率模型的算法,能够快速计算出转录本的表达水平,并允许处理带有复杂可变剪接事件的数据集。
亮点功能:
- 基于概率的快速定量:salmon使用了先进的概率模型,使其在处理速度和准确性上具有显著优势,可以快速处理大量数据,同时保证高精度。
- 实时定量:salmon支持实时计算基因表达水平,允许用户在数据处理的过程中不断更新和调整分析结果 (GitHub)。
- 灵活的输入和输出选项:salmon支持多种输入格式,并提供详细的输出结果,包括基因和转录本层次上的表达估算值 (GitHub)。
code:https://github.com/COMBINE-lab/salmon
适用场景: salmon特别适合需要快速定量和处理复杂数据集的研究场景,如复杂疾病研究、发育生物学研究和环境响应研究。
Top5:kallisto
kallisto是一款自2015年问世以来在生物信息学界广受欢迎的RNA-seq数据定量工具,以其极高的计算效率和对大规模数据集的适用性而著称。
工具简介: kallisto是一款用于快速、准确估算基因表达水平的工具,基于创新的伪比对算法,可以在不需要完全比对的情况下,迅速计算出转录本的丰度。其速度和资源占用都显著优于传统的比对方法。
亮点功能:
- 伪比对算法:kallisto利用伪比对(pseudoalignment)技术,通过直接将RNA-seq读段与转录本索引进行比较,大幅减少了计算时间。
- 高效的内存使用:kallisto能够在较低的内存占用下处理大量数据,这使得它非常适合处理包含上百万读段的大规模RNA-seq实验 (GitHub)。
- 精确的转录本定量:尽管使用了伪比对技术,kallisto依然能够提供与传统比对方法相媲美的定量精度,非常适合用于基因表达谱分析 (GitHub)。
Code: https://github.com/pachterlab/kallisto
适用场景: kallisto非常适合基因表达分析,尤其是在需要处理大规模数据集或有限计算资源的情况下。它在癌症研究、发育生物学和其他需要高效RNA-seq数据处理的研究中被广泛应用。
排名评价
值得注意的是,STAR和Seurat的引用量遥遥领先,分别达到了惊人的3万多次和2.8万多次。STAR的广泛应用不仅限于单细胞数据,还包括常规的RNA-seq数据处理,这解释了其超高的引用量。Seurat则凭借其出色的性能,成为单细胞转录组数据分析的首选工具,尽管在处理大规模数据时可能面临计算效率的挑战。
对于Python爱好者关心的Scanpy排名和介绍,我们将在下期揭晓~
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-08-27,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号