DFT计算和MD模拟技术在水系电池中的应用-测试GO
原创

1. 界面反应机制与稳定性验证
CEI/SEI形成机制
DFT和MD被用于解析阴极-电解液界面(CEI)和固体电解质界面(SEI)的原子级反应路径。例如,在锂离子电池中,DFT计算揭示了LiF在SEI中的优先形成机制,其低扩散能垒(约0.68 eV)有利于离子传输。MD模拟进一步验证了水分子参与界面分解的过程,解释了氢析出反应(HER)的触发条件。
高电压界面稳定性
针对高电压水系电池(如>2.5 V窗口),DFT计算预测了电解液成分(如高浓度LiTFSI)的氧化分解路径,并通过MD验证了"盐包水"电解液中阴离子富集层对抑制氧析出反应(OER)的作用。
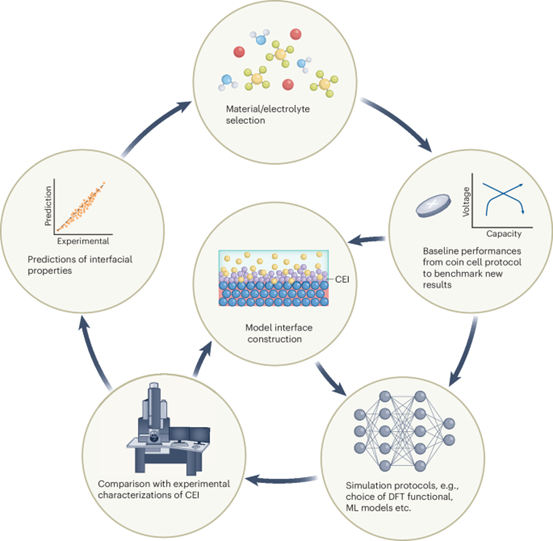
CEI研究的多尺度模拟工作流
CEI研究的多尺度模拟工作流
2. 离子输运动力学与溶剂化结构
离子迁移能垒计算
DFT计算量化了多价金属离子(如Zn²⁺、Al³⁺)在水溶液中的脱溶剂化能垒。Al³⁺因高水合能(~4660 kJ/mol)导致缓慢动力学,MD模拟显示其水合壳层结构需通过电解液设计(如添加Cl⁻)削弱。
溶剂化结构动态演化
MD模拟揭示了"盐包水"电解液中离子对(CIPs)和聚集体(AGGs)的形成规律。例如,21 m LiTFSI电解液中,Li⁺第一溶剂化壳层中自由水比例降至<10%,显著拓宽电化学窗口至3.0 V。
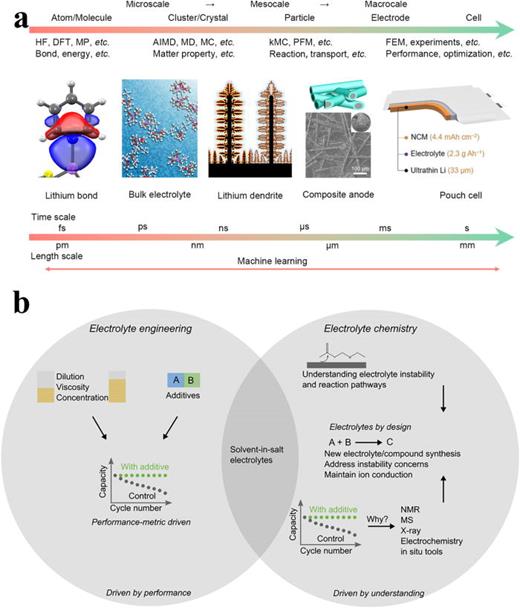
离子输运特性的多尺度建模
离子输运特性的多尺度建模
3. 电极材料设计与性能优化
材料缺陷与掺杂效应
DFT计算预测了锰基阴极材料中氧空位对Zn²⁺嵌入能垒的影响,揭示了Mn³⁺/Mn⁴⁺氧化还原电位偏移机制 。通过MD验证了Na⁺预嵌入调控层间距(增加~0.3 Å)提升Zn²⁺扩散系数的路径。
界面修饰策略验证
针对锌负极枝晶问题,DFT计算证明碳基材料(如石墨烯)的锌亲和性(吸附能<-0.5 eV)可诱导均匀成核;MD模拟进一步显示表面涂层(如MOF衍生碳)能调节Zn²⁺通量分布。
4. 电解液设计中的关键问题
添加剂作用机制
DFT计算筛选了抑制HER的添加剂(如Na₂SO₄、有机分子),通过H₂O分子轨道能级与添加剂LUMO能级匹配度预测还原稳定性。MD模拟显示静电屏蔽添加剂(如Bi³⁺)通过吸附层排斥游离H⁺,减少界面副反应。
离子选择性传输
在双离子电池中,MD模拟证实阴离子交换膜的孔径(<0.6 nm)可调控阴/阳离子选择性渗透率(如SO₄²⁻/Zn²⁺分离效率>90%)。
技术局限性
时间与空间尺度的分离 MD模拟通常限于纳米级模型(<10 nm)和纳秒级动态过程,难以捕捉宏观尺度下的电极退化(如循环百次后的相变)。
力场精度依赖 水溶液中的离子-溶剂作用力(如Zn-H₂O)缺乏普适力场,需通过DFT数据矫正。
界面模型的简化 实际固液界面的粗糙度及缺陷难以在DFT表面模型中复现,影响电荷分布预测的准确性。
未来方向
DFT与MD的协同可向多尺度耦合(如DFT/MD-AI联用)拓展。例如,通过机器学习势函数(ML-FF)将DFT精度与MD尺度结合,用于高通量筛选固态电解质(如LATP)的界面钝化层组分;或预测新型导电MOF材料在水系锌电池中的拓扑效应。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号