空间转录组学: 局部异常检测


引言
本系列讲解 空间转录组学 (Spatial Transcriptomics) 相关基础知识与数据分析教程[1],持续更新,欢迎关注,转发,文末有交流群!
局部异常检测
单细胞与空间转录组学中,全局异常检测的一个关键假设是:QC 指标与自然生物学相互独立。否则,天然具有低文库大小或较高线粒体比例的细胞或 spot 更可能被当作异常值移除。对于 snRNA-seq,这一假设很少或仅轻微被违背,对下游分析的影响可忽略不计。然而,如上所示,在基于测序的 ST 中,由于每个 spot 所采样组织的生物学异质性更高,该假设更常被违背。
解决此问题的一种策略是在其局部生物学邻域内寻找异常值。在此,我们将使用 SpotSweeper Bioconductor 包中的 localOutliers() 函数实现局部异常检测。该函数通过比较每个 spot 与其最近邻的检测到的独特基因数、总文库大小和线粒体百分比来识别局部异常值。默认情况下,localOutliers() 使用 k = 36 个最近邻,这对应于 Visium 六边形 spot 布局中的三阶邻域(即围绕每个 spot 的三层同心邻域)。然而,对于使用方形网格布局的基于测序的方法(例如 STOmics),三阶邻域将为 k = 48。
与上文使用的自适应阈值类似,这些方法假设正态分布,因此我们将对总 counts 的对数转换和检测到的基因数的对数转换使用。我们不会对线粒体百分比进行对数转换,因为其倾向于遵循正态分布。
# library size
spe <- localOutliers(spe, metric = "sum", direction = "lower", log = TRUE)
# unique genes
spe <- localOutliers(spe, metric = "detected", direction = "lower", log = TRUE)
# mitochondrial percent
spe <- localOutliers(spe, metric = "subsets_mito_percent",
direction = "higher", log = FALSE)
与 scater 的 addPerCellQC() 函数类似,localOutliers() 函数会向 SpatialExperiment 对象的 colData slot 添加若干列。X_outlier 列包含一个逻辑向量,用于指示该 spot 在对应指标上是否为异常值;X_z 列返回该指标的局部 z-score 转换后的 QC 指标。如果 log = TRUE,还会额外生成一个 X_log 列,返回经过对数转换后的指标值。
随后,我们可以通过 spot 图(spot plots)对这些 QC 指标进行可视化,以直观确认检测到的局部异常值确实为异常。为此,我们将把输出的 log2 转换数据(或未经转换的线粒体比例)与检测到的局部异常值并置展示。
# spot plot of log-transformed library size
p1 <-
plotCoords(spe, annotate="sum_log") +
ggtitle("log2(Library Size)")
p2 <-
plotObsQC(spe, plot_type = "spot", in_tissue = "in_tissue",
annotate = "sum_outliers", point_size = 0.2) +
ggtitle("Local Outliers (Library Size)")
# spot plot of log-transformed detected genes
p3 <-
plotCoords(spe, annotate = "detected_log") +
ggtitle("log2(Detected)")
p4 <-
plotObsQC(spe, plot_type = "spot", in_tissue = "in_tissue",
annotate = "detected_outliers", point_size = 0.2) +
ggtitle("Local Outliers (Detected)")
# spot plot of mitochondrial proportion
p5 <-
plotCoords(spe, annotate = "subsets_mito_percent") +
ggtitle("Mito Proportion")
p6 <-
plotObsQC(spe, plot_type = "spot", in_tissue = "in_tissue",
annotate = "subsets_mito_percent_outliers", point_size = 0.2) +
ggtitle("Local Outliers (Mito Prop)")
# plot using patchwork
(p1 / p2) | (p3 / p4) | (p5 / p6)
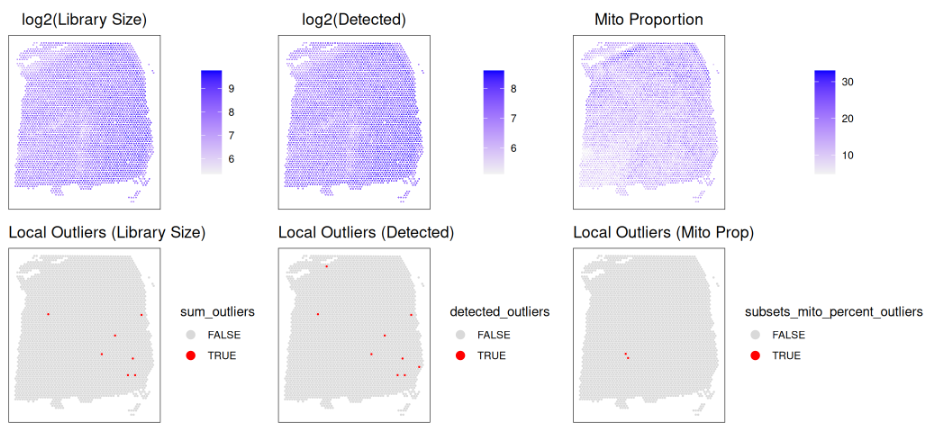
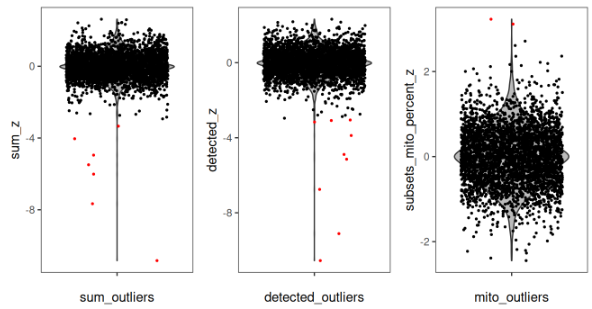
在对数转换后的文库大小和检测到的基因数上尤其明显,在组织区域的右下角存在清晰的异常值。我们还可以看到,这些异常 spot 在底部一行被成功识别。这种“目测检验”是一个良好的诊断方法,用于确认局部异常值被准确检测。另一种做法是,通过小提琴图对每个 spot 的 z-score 转换后的指标进行可视化,从而展示被检测为异常值的 spot。
# z-transformed library size and outliers
p1 <-
plotObsQC(spe, plot_type = "violin", x_metric = "sum_z",
annotate = "sum_outliers", point_size = 0.5) +
xlab("sum_outliers")
# z-transformed detected genes and outliers
p2 <-
plotObsQC(spe, plot_type = "violin", x_metric = "detected_z",
annotate = "detected_outliers", point_size = 0.5) +
xlab("detected_outliers")
# z-transformed mito percent and outliers
p3 <-
plotObsQC(spe, plot_type = "violin", x_metric = "subsets_mito_percent_z",
annotate = "subsets_mito_percent_outliers", point_size = 0.5) +
xlab("mito_outliers")
# plot using patchwork
p1 | p2 | p3
未完待续,欢迎关注!
Reference
[1]
Ref: https://lmweber.org/OSTA/
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-08-19,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号