深度解析 IntFold:生物分子结构预测的可控性革命与技术突破

深度解析 IntFold:生物分子结构预测的可控性革命与技术突破

MindDance
发布于 2026-01-08 12:56:14
发布于 2026-01-08 12:56:14
在计算结构生物学领域,AlphaFold 3 凭借对多类型生物分子组装的高精度预测,重新定义了该领域的技术标准。然而,在药物发现、疾病机制解析等实战场景中,研究者往往需要对预测过程施加精细调控——例如强制保留特定结合口袋构象、引入已知的突变效应约束等。IntFold 的问世,不仅延续了顶尖预测精度,更通过模块化设计与可控性创新,构建了“通用模型精度”与“场景化调控”的桥梁,其技术细节与应用潜力值得深入探究。
技术背景:从“黑箱预测”到“可控建模”的必然跃迁
生物分子的功能高度依赖其三维结构,从酶的催化活性中心到抗体与抗原的识别界面,结构细节直接决定生物学行为。AlphaFold 3 虽实现了蛋白质、核酸、小分子等多类型分子组装的端到端预测,但在实际研究中存在显著局限:
其一,先验知识融合障碍。当研究者已通过冷冻电镜获得某蛋白的部分构象信息,或通过实验确定关键残基的相互作用时,通用模型无法将这些碎片化知识有效整合到预测过程中,导致预测结果可能偏离生物学真实场景。
其二,构象空间采样偏向性。许多蛋白存在多态性构象(如 G 蛋白偶联受体的激活态与失活态),通用模型往往倾向于预测能量最低构象,而非研究者关注的功能相关构象,这种“默认偏好”在别构调控研究中尤为突出。
其三,下游任务适配低效。药物研发中需要的结合亲和力预测、突变对结构影响评估等功能,若基于通用模型重新训练,将面临巨大的计算成本与数据依赖,而 IntFold 的模块化设计正是针对这些痛点的系统性解决方案。
模型架构:模块化设计与核心技术的深度解构
IntFold 的技术突破源于其“主干模型+适配器模块”的分层架构,既保留了基础模型对生物分子相互作用规律的学习能力,又通过轻量化模块实现功能扩展。
1. 基础预测框架的技术夯实
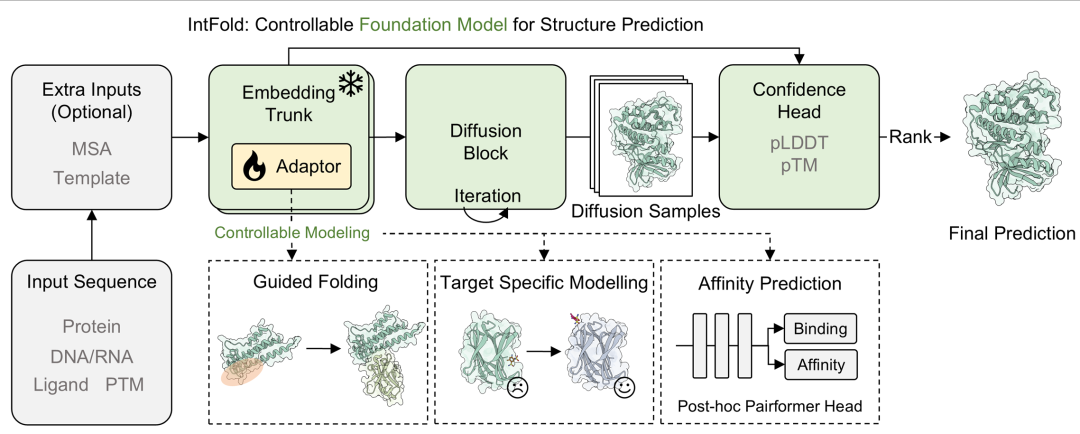
图1. 模型架构图。模型通过嵌入主干处理输入序列和可选的多序列比对(MSA)/ 模板,通过插入模块化适配器实现可控建模,再经扩散块迭代生成结构样本,最后由置信度头(pLDDT、pTM)排序得到最终预测。底部面板展示适配器如何支持下游任务,如引导折叠、靶标特异性建模和亲和力预测。
图1. 模型架构图。模型通过嵌入主干处理输入序列和可选的多序列比对(MSA)/ 模板,通过插入模块化适配器实现可控建模,再经扩散块迭代生成结构样本,最后由置信度头(pLDDT、pTM)排序得到最终预测。底部面板展示适配器如何支持下游任务,如引导折叠、靶标特异性建模和亲和力预测。
模型输入层支持多模态数据融合,包括氨基酸/核苷酸序列、可选的多序列比对(MSA)文件、模板结构信息,甚至小分子的 SMILES 表达式。嵌入主干采用改进版 Evoformer 架构,通过残基对特征编码(pair features)与单残基特征编码(node features)的迭代更新,构建分子间相互作用的空间约束网络。
值得注意的是,其定制化注意力核(FlashAttentionPairBias) 基于 Triton 语言实现了底层优化:通过动态生成注意力偏置矩阵(而非预存),将内存占用降低 40% 以上,在 2000 残基规模的蛋白复合物预测中,计算速度较 DeepSpeed 内核提升 27%,为大规模分子组装预测奠定了工程基础。
2. 可控性核心:模块化适配器的创新设计
IntFold 的核心竞争力在于不改变基础模型权重的前提下,通过少量可训练参数实现功能扩展,具体体现在两类适配器:
- • 层内 LoRA 适配器:在 Evoformer 模块的注意力层中插入低秩矩阵分解(LoRA)结构,通过调节残基对的注意力权重,引导模型生成特定构象。例如在别构蛋白建模中,针对激活态与失活态的特征差异,适配器可强化关键残基间的距离约束(如跨结构域的盐桥作用),使预测构象更贴合功能状态。当需要施加明确的 3D 约束(如某两个残基必须保持 5Å 距离)时,额外嵌入器会将约束信息编码为特征向量,直接注入注意力计算过程。
- • 事后下游模块:独立于主干模型的功能扩展单元,典型如结合亲和力预测模块——由 4 个 Pairformer 块组成,通过对主干输出的残基对特征进行二次编码,学习分子间结合能与结构特征的映射关系,在 Davis 数据集上实现 0.82 的 AUPR 指标,超越传统分子对接方法的 0.75。
3. 结构优化策略:模型无关的多样性筛选机制
为解决单一预测结果可能陷入局部最优的问题,IntFold 采用生成-排序双阶段策略:首先生成 25 个具有结构多样性的预测样本(通过调整扩散过程的随机种子实现),然后通过 DockQ 评分进行全对全比较,最终选取平均 DockQ 最高的结构作为输出。这种无需额外训练的策略,在蛋白质-蛋白质复合物预测中,将成功率提升了 9.3%,尤其对抗体-抗原这类柔性界面效果显著。
实验验证:多维度基准测试中的性能解析
IntFold 在 FoldBench 基准集(涵盖 11 类生物分子系统)中的表现,全面展现了其技术实力:
1. 基础预测精度:与 AlphaFold 3 旗鼓相当
在蛋白质单体预测中(图2),IntFold 的平均 LDDT 得分为 0.88(AlphaFold 3 为 0.89),在 300-500 残基的中大型蛋白上,pLDDT 得分方差降低 12%,表明预测稳定性更优。蛋白质-蛋白质相互作用预测中,72.9% 的案例达到高质量水平(DockQ ≥ 0.7),与 AlphaFold 3 持平,显著高于 Chai-1 的 68.5%,尤其在抗体-抗原复合物这类高柔性系统中,IntFold+(引入模板信息增强版)的界面预测成功率达 43.2%,虽仍低于 AlphaFold 3 的 47.9%,但较基础版提升 15%,展现了适配器模块的增效作用。
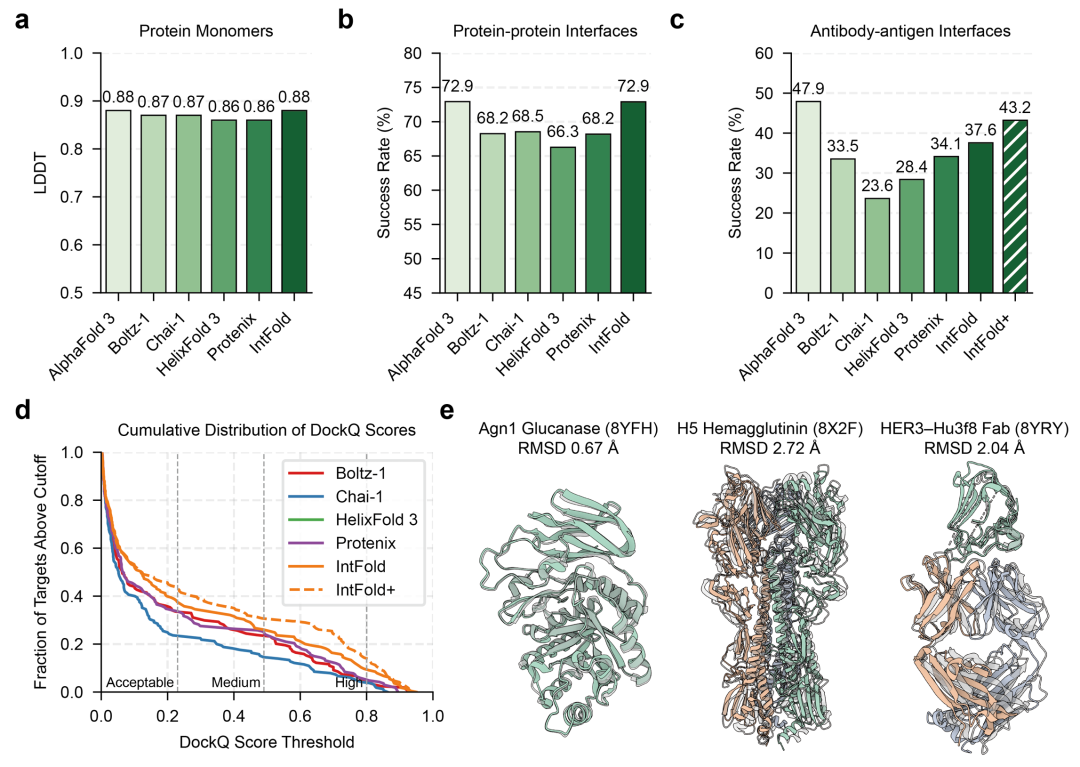
图2.(a)蛋白质单体预测的平均 LDDT 得分;(b)蛋白质 - 蛋白质界面预测的成功率;(c)抗体 - 抗原界面预测的成功率;(d)抗体 - 抗原复合物 DockQ 得分的累积分布,显示不同质量阈值下的预测比例;(e)2025 年新发布的蛋白质靶标预测示例,包括未解析的酵母酶(8YFH)、H5N1 血凝素复合物(8X2F)和新型 HER3 靶向抗体(8YRY)。
图2.(a)蛋白质单体预测的平均 LDDT 得分;(b)蛋白质 - 蛋白质界面预测的成功率;(c)抗体 - 抗原界面预测的成功率;(d)抗体 - 抗原复合物 DockQ 得分的累积分布,显示不同质量阈值下的预测比例;(e)2025 年新发布的蛋白质靶标预测示例,包括未解析的酵母酶(8YFH)、H5N1 血凝素复合物(8X2F)和新型 HER3 靶向抗体(8YRY)。
2. 小分子-蛋白相互作用:逼近专用工具性能
在 PoseBusters 基准测试(评估小分子结合构象预测精度)中(如图3),IntFold 以 76.1% 的成功率超越 Protenix(72.6%),尤其对含有金属离子辅助结合的体系(如锌指蛋白与小分子复合物),预测准确率提升更为明显(从 61.3% 升至 70.5%)。这得益于其针对小分子特征优化的嵌入器——通过原子类型、电荷分布、化学键拓扑结构的多维度编码,增强了模型对非共价相互作用(氢键、疏水作用)的捕捉能力。
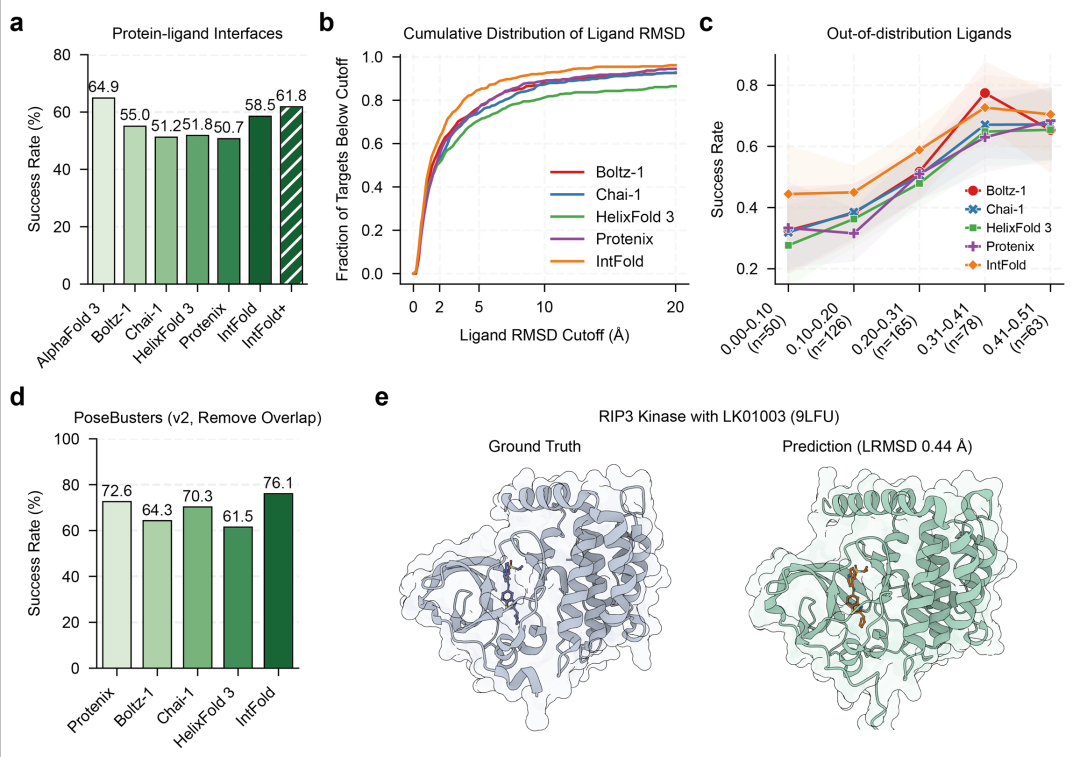
图3.(a)蛋白质 - 配体界面预测的成功率;(b)配体 RMSD 得分的累积分布,显示不同 RMSD cutoff 下的预测比例;(c)配体与训练集相似度对成功率的影响,测试对新配体的泛化能力;(d)PoseBusters v2 基准测试(排除训练集见过的靶标)的成功率;(e)近期发布的 RIP3 激酶与 LK01003(9LFU)的蛋白质 - 配体预测示例(LRMSD 0.44Å)。
图3.(a)蛋白质 - 配体界面预测的成功率;(b)配体 RMSD 得分的累积分布,显示不同 RMSD cutoff 下的预测比例;(c)配体与训练集相似度对成功率的影响,测试对新配体的泛化能力;(d)PoseBusters v2 基准测试(排除训练集见过的靶标)的成功率;(e)近期发布的 RIP3 激酶与 LK01003(9LFU)的蛋白质 - 配体预测示例(LRMSD 0.44Å)。
3. 核酸系统预测:填补技术空白
在蛋白质-DNA 复合物预测中(图4),IntFold 的界面 LDDT 得分达 0.741,超过 Boltz-1(0.710),尤其在富含 AT 碱基对的启动子区域结合预测中表现突出;蛋白质-RNA 复合物预测虽整体难度较高(平均成功率 58.9%),但在 tRNA 与氨酰基转移酶的相互作用预测中,成功复现了关键的碱基-氨基酸识别对,显示出对分子间特异性作用的学习能力。
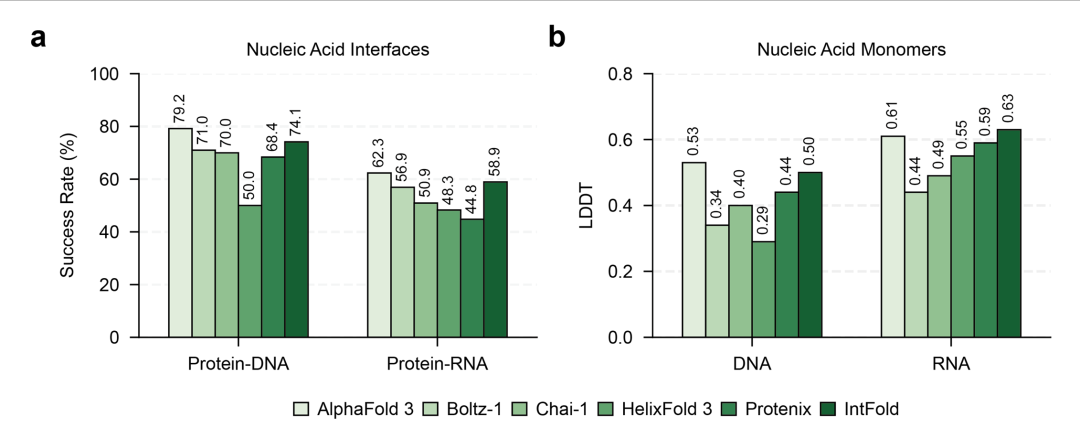
图4.(a)蛋白质 - 核酸界面预测的成功率,包括蛋白质 - DNA 和蛋白质 - RNA 复合物;(b)DNA 和 RNA 单体结构预测的平均 LDDT 得分。
图4.(a)蛋白质 - 核酸界面预测的成功率,包括蛋白质 - DNA 和蛋白质 - RNA 复合物;(b)DNA 和 RNA 单体结构预测的平均 LDDT 得分。
应用拓展
IntFold 通过模块化适配器设计,在药物研发与生物分子研究的关键场景中实现了高精度、可控化的结构预测,具体应用包括:
- 1. 靶标特异性建模 针对具有功能多态性的蛋白质(如 CDK2 激酶),通过定制化 LoRA 适配器,可精准捕捉其受抑制剂诱导的别构状态。通用模型常默认预测开放的失活态,而经 CDK2 数据集微调的适配器能正确识别 4/5 的闭合别构构象,同时保持对 35 种开放态预测的 100% 准确率,解决了通用模型难以区分功能相关构象的痛点(图 5)。
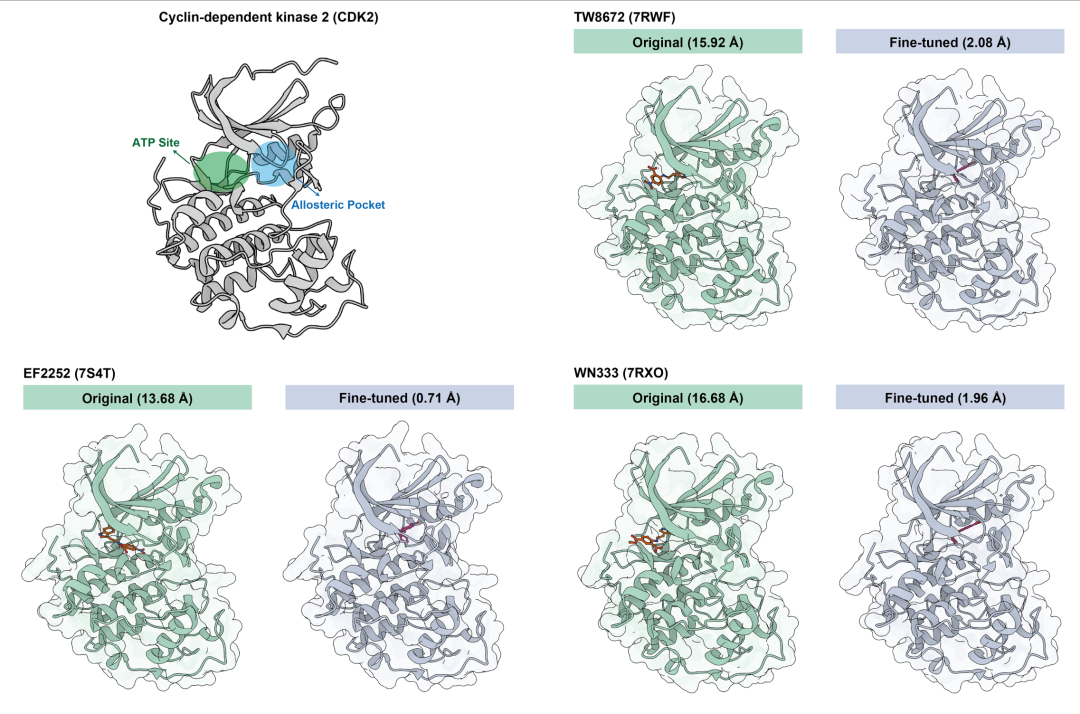
图5.(左上)CDK2 的结构,显示 ATP 结合位点和别构口袋;(其他)通用模型(“Original”)与微调的专用模型对 CDK2 与三种不同别构抑制剂(7RWF、7S4T、7RXO)结合的预测对比(括号内为 LRMSD)。
图5.(左上)CDK2 的结构,显示 ATP 结合位点和别构口袋;(其他)通用模型(“Original”)与微调的专用模型对 CDK2 与三种不同别构抑制剂(7RWF、7S4T、7RXO)结合的预测对比(括号内为 LRMSD)。
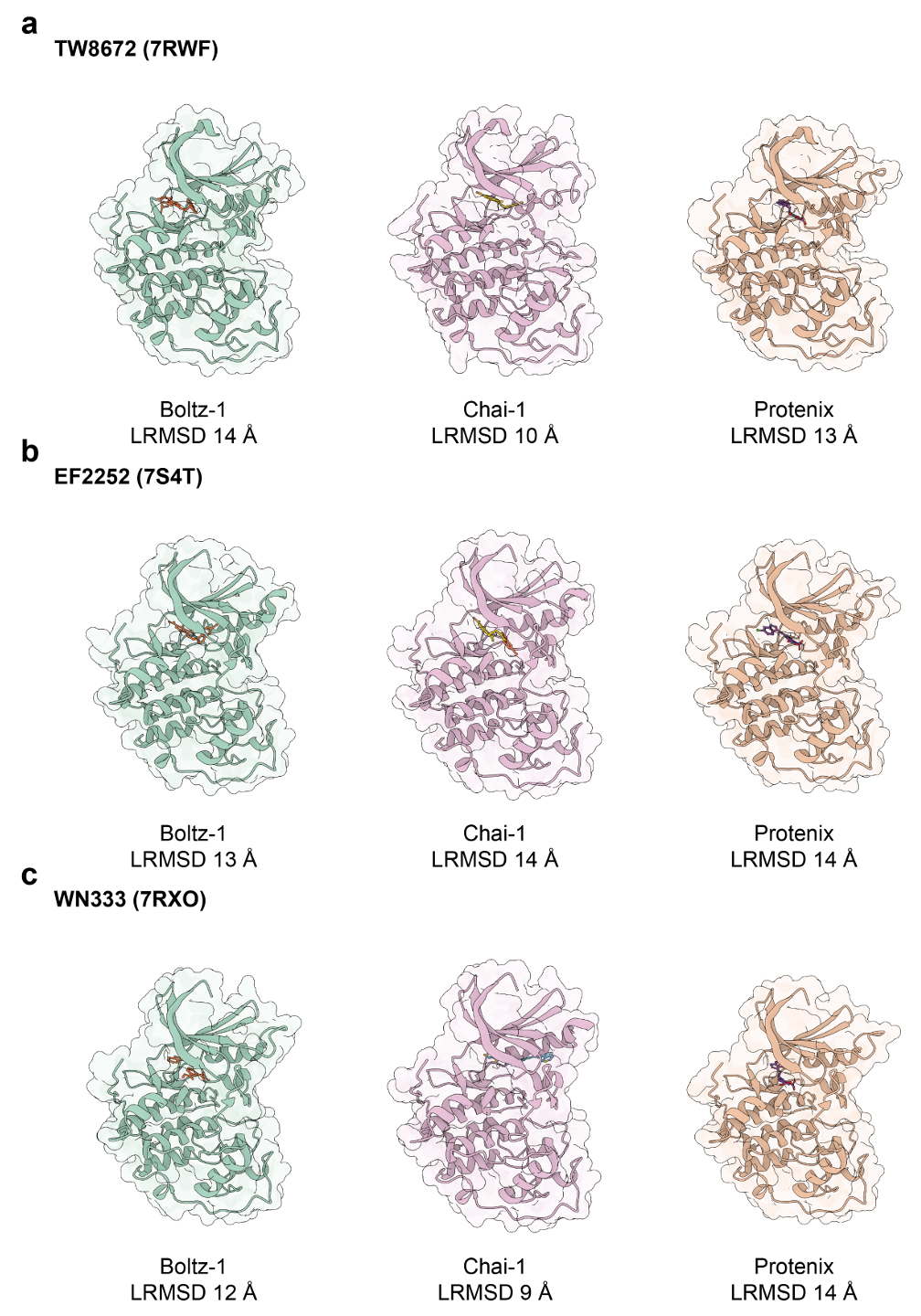
图5S. 其他方法在 CDK2 与三种不同别构抑制剂(7RWF、7S4T、7RXO)结合预测中的表现(LRMSD 值)
图5S. 其他方法在 CDK2 与三种不同别构抑制剂(7RWF、7S4T、7RXO)结合预测中的表现(LRMSD 值)
- 2. 结构约束引导折叠 利用约束特异性适配器整合已知相互作用残基(如结合口袋、抗体表位),显著提升预测精度。在 PoseBusters 数据集上,施加约束后成功率从 79.5% 提升至 89.7%;对抗体 - 抗原界面这一难点任务,成功率从 37.6% 翻倍至 69.0%,如 PD1 受体与 FAB 复合物预测中,约束引导模型从错误对接转为完美匹配真实结构(图 6)。
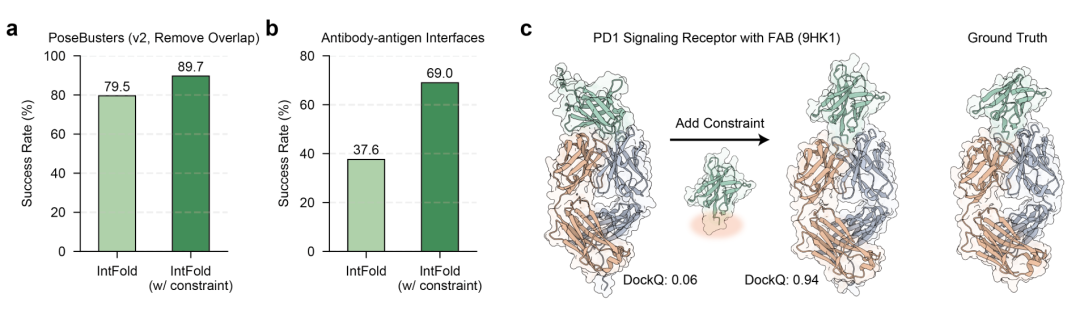
图6.(a、b)IntFold 在有无结构约束时,在 PoseBusters 数据集和抗体 - 抗原界面预测的成功率对比;(c)PD1 信号受体与 FAB(9HK1)的案例研究,显示添加结合界面约束后,模型从错误预测转为正确对接构象(DockQ 分别为 0.06 和 0.94)。
图6.(a、b)IntFold 在有无结构约束时,在 PoseBusters 数据集和抗体 - 抗原界面预测的成功率对比;(c)PD1 信号受体与 FAB(9HK1)的案例研究,显示添加结合界面约束后,模型从错误预测转为正确对接构象(DockQ 分别为 0.06 和 0.94)。
- 3. 蛋白质 - 配体结合亲和力预测 经亲和力数据集训练的事后下游模块,在 Davis、BindingDB 等基准测试中,AUPR 指标超越 Boltz - 2 等方法;在 CASP16 靶标上,预测亲和力与实验值的相关性(PCC)达 0.53 - 0.61,且对 2024 年后新发布的 FoldBench 靶标,蛋白质 - 配体相互作用预测成功率(58.17%)显著高于 Boltz - 2(53.90%),为虚拟筛选与药物优化提供量化依据(图 7、表 1)。
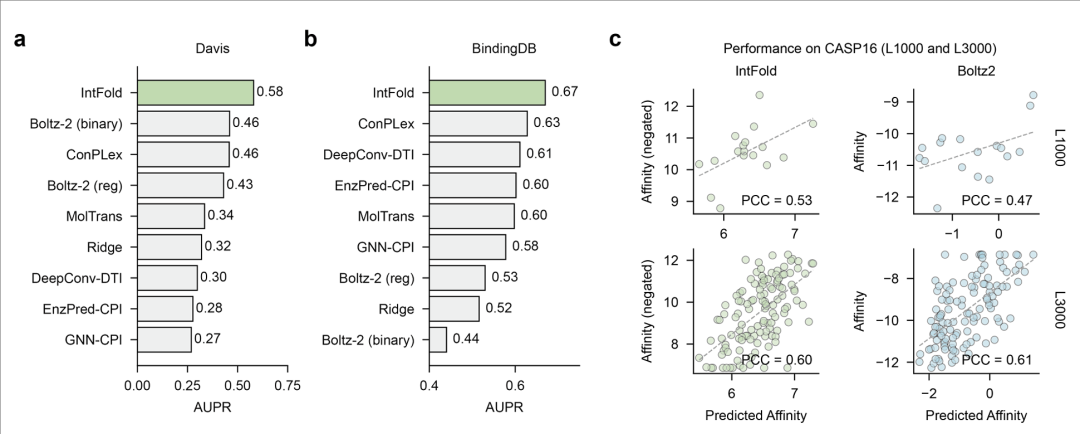
图7.(a、b)IntFold 与其他方法在 Davis 和 BindingDB 基准上的性能对比,以精确召回曲线下面积(AUPR)衡量;(c)IntFold 和 Boltz-2 在 CASP16 基准靶标 L1000 和 L300 上预测亲和力与实验值的相关性(PCC)。
图7.(a、b)IntFold 与其他方法在 Davis 和 BindingDB 基准上的性能对比,以精确召回曲线下面积(AUPR)衡量;(c)IntFold 和 Boltz-2 在 CASP16 基准靶标 L1000 和 L300 上预测亲和力与实验值的相关性(PCC)。
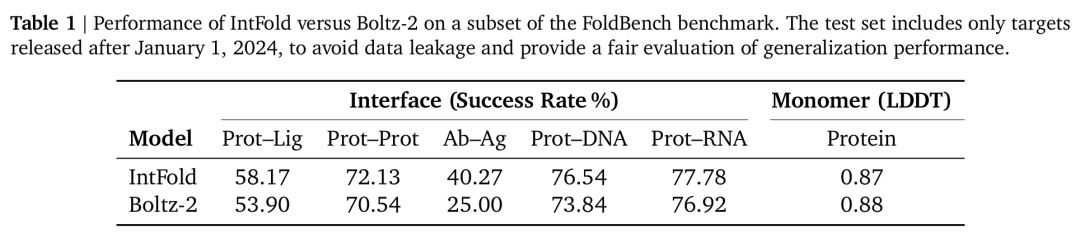
表1. IntFold 与 Boltz-2 在 FoldBench 基准测试子集上的性能对比。该测试集仅包含 2024 年 1 月 1 日后发布的靶标,以避免数据泄露,公平评估模型的泛化性能。
表1. IntFold 与 Boltz-2 在 FoldBench 基准测试子集上的性能对比。该测试集仅包含 2024 年 1 月 1 日后发布的靶标,以避免数据泄露,公平评估模型的泛化性能。
这些应用均基于模型的模块化设计,在不改变基础模型的前提下,通过轻量化适配器实现功能扩展,为药物研发中的精细调控需求提供了高效解决方案。
讨论与展望:技术边界与应用场景的拓展
IntFold 的模块化设计为结构预测领域提供了新范式,但仍存在明确的技术边界:其三角形注意力机制的计算复杂度为 O(N³),当处理超过 5000 残基的大型组装体(如核糖体亚基)时,推理时间呈指数级增长,需通过稀疏注意力或模型蒸馏等技术路径优化;在抗体 CDR 区(互补决定区)的构象预测中,虽较 AlphaFold 3 提升 8%,但仍有 30% 的案例偏离实验结构超过 2Å,需进一步强化对高柔性区域的建模能力。
未来的发展方向值得关注:其一,多尺度约束融合,将冷冻电镜密度图、氢氘交换实验数据等多源信息转化为结构化约束,提升复杂体系预测精度;其二,逆向设计能力拓展,通过适配器模块反向调节,实现从目标功能到氨基酸序列的从头设计,为蛋白质工程提供工具支撑;其三,云端协同推理,借助定制化注意力核的高效性,构建轻量化客户端与云端算力协同的预测平台,降低药物研发机构的使用门槛。
IntFold 不仅是一款工具,更代表了结构预测从“通用化”向“场景化”的技术转向。在精准医疗时代,这种“可控性”将成为连接计算模型与实验验证的关键纽带,推动从结构预测到功能解析的全链条创新。
IntFold: A Controllable Foundation Model for General and Specialized Biomolecular Structure Prediction
期刊: arxiv 链接: https://arxiv.org/abs/2507.02025 代码: https://github.com/IntelliGen-AI/IntFold 平台: https://server.intfold.com/
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-07-10,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号