RNA速率分析中遇到的问题以及debug纪实
原创

根据之前的推文,我们获得了全部样本合并后的loom文件,但在我使用scvelo进行分析时惊讶的发现,将单细胞矩阵和loom文件合并后细胞数从11万减少到了5万。
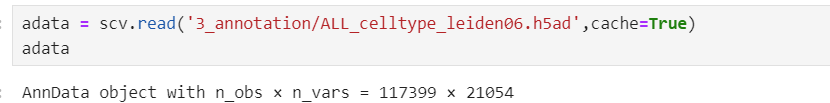

这是合并前的两个文件,细胞数都在10万以上
当我使用scvelo将这两个文件合并后惊讶的发现细胞数只剩下了5万
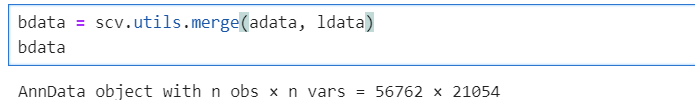
为了探究导致这一问题的原因,我详细检查了数据结构。
首先,基因数量没有变化,暂时不管var.names,细胞数发生了如此明显的改变,我需要先检查adata.obs与ldata.obs。
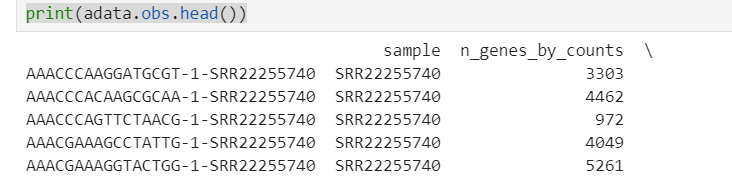
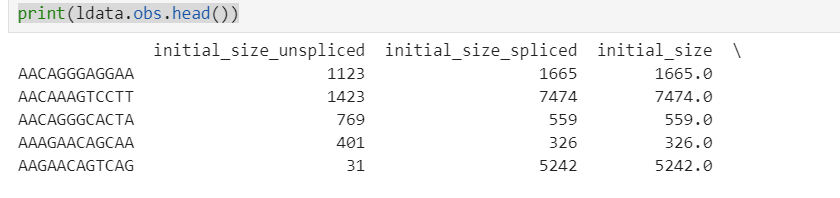
可以看到,细胞id发生了改变,随后我检查了cellranger输出结果中的barcode
zless outs/filtered_feature_bc_matrix/barcodes.tsv.gz | head -n 10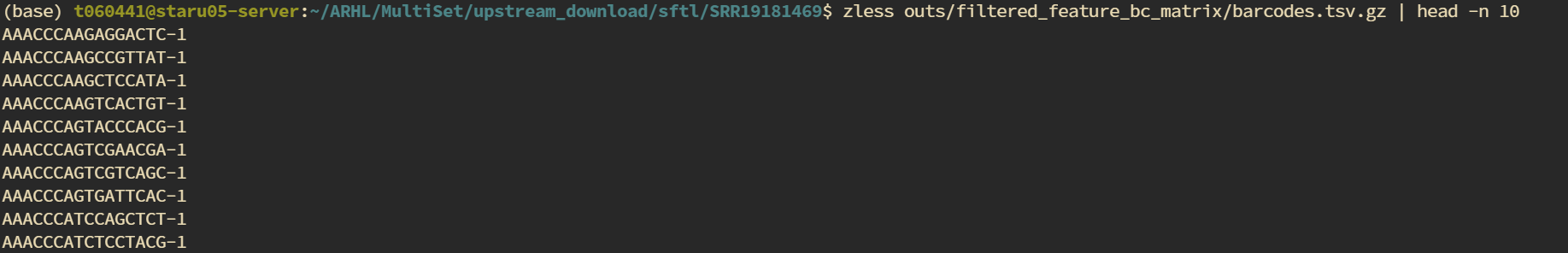
可以看到adata中的barcode格式与cellranger输出的格式一致,只是后面添加了样本信息
接下来,问题问题直接指向ldata这个loom文件。我检查了velocyto的源码,发现他在运行过程中不会导致barcode格式发生如此严重的改变。
首先,在run10x这一函数中没有对barcode进行任何改动。
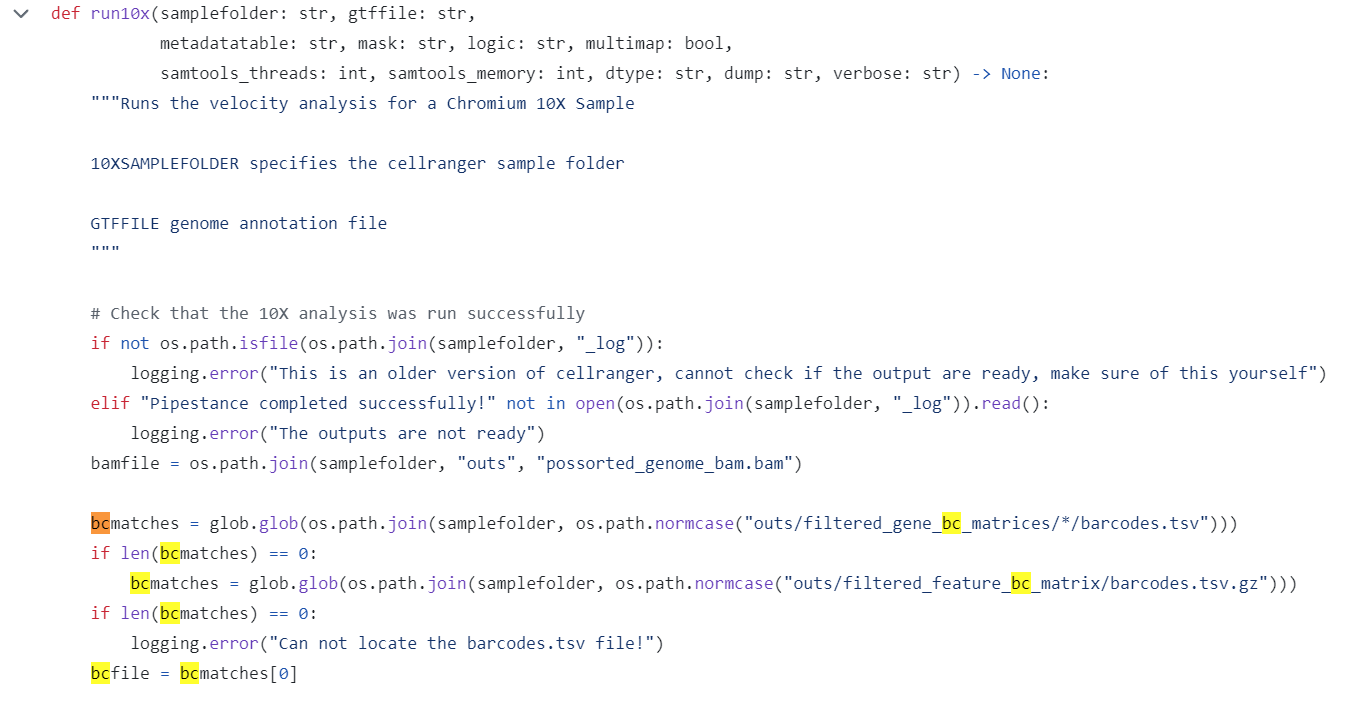
这一步仅仅是读取了cellranger输出结果的barcode文件
源码的后半部分将bcfile传递给了_run函数
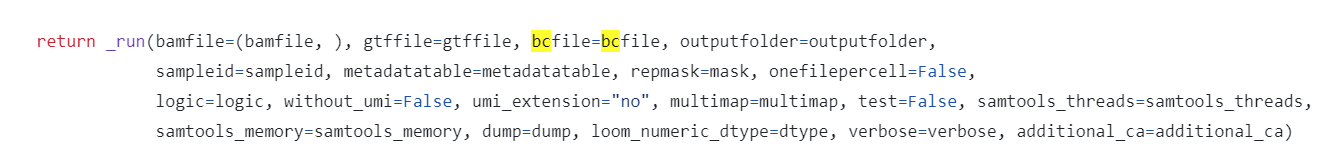
这一参数才真正使用到了run10x中的输入信息,并将上一步得到的读取自barcode.tsv的bcfile继承
打开_run.py源码,直接定位到bcfile,我们可以发现,这部分源码唯一的改动就是对-1前没有重复的barcode加上一个‘x’

为了验证我的结论,我读取了一个合并前的loom文件。发现细胞id果然没有发生如此大的改变。barcode前添加了sample id,barcode结尾的-1被替换成了'x'
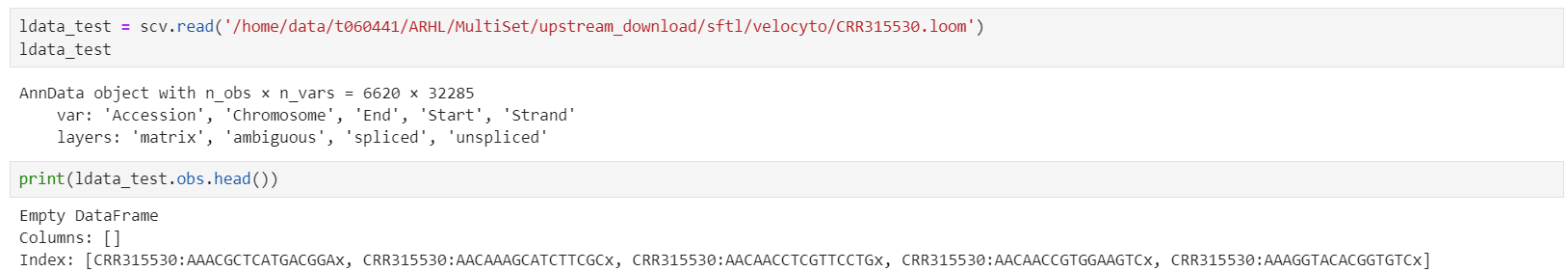
接下来检查合并loom文件这一步。
我用来合并的代码是直接复制于velocyto官网

使用loompy中的combine函数完成,但在我们查看源码时发现combine函数及其调用的add_loom中均没有直接改变(源码过长,这里不截图了)。那么现在唯一不同之处在于loom文件的barcode命名格式与scanpy等软件不同,我们在scvelo的merge函数中发现了如下内容:

这里检查了adata与ldata的obs_name是否相同
所以问题的根源就在run.py这里,我们直接修改ldata的barcode再进行merge,根据velocyto源码中的内容:首先将sampe id添加于barcode之前,然后检查16位的barcode是否有重复,如果有重复把-1替换为x,如果没有就保留-1。我们直接粗暴还原
import pandas as pd
# 定义修改函数
def modify_obs_name(obs_name):
prefix, barcode = obs_name.split(':')
new_barcode = barcode[:16] + '-1' + barcode[17:]
return f"{new_barcode}-{prefix}"
# 应用修改函数到所有的obs名称
ldata.obs_names = [modify_obs_name(obs_name) for obs_name in ldata.obs_names]
# 检查修改结果
print(ldata.obs.head())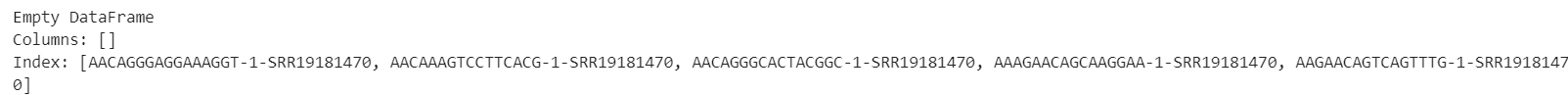
格式已经一样了
接下来判断一下adata中是否存在这些名字
# 判断 adata.obs_names 是否全部存在于 ldata.obs_names 中
all_in = adata.obs_names.isin(ldata.obs_names).all()
# 输出结果
if all_in:
print("adata.obs_names 全部存在于 ldata.obs_names 中。")
else:
print("adata.obs_names 中有一些不在 ldata.obs_names 中。")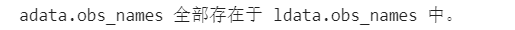
搞定
解决掉上述问题之后,重新执行scv.utils.merge,问题已经解决!

可以看到现在合并后的单细胞矩阵保留全部质控后的细胞(细胞数与adata相同)
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号