Visium HD 空间转录组分析探索之--基础分析
原创
Visium HD 空间转录组分析探索之--基础分析
原创
生信大杂烩
发布于 2025-05-29 21:23:09
发布于 2025-05-29 21:23:09
上节我们完成了Space Ranger分析,在输出结果binned_outputs文件夹内默认生成了2um, 8um, 16um bin的结果,这节我们使用10x文章中推荐的8x8um bin结果进行后续分析。为了演示,我们取P1_CRC样本数据进行后续分析(多样本合并一般都需要进行去批次处理,如果有需要,后续我们会继续分享多样本合并分析步骤)。
主要步骤:
1.读取8um bin结果;
2.数据质控;
3.细胞过滤;
4.降维聚类;
5.特征基因筛选
环境加载
import osimport numpy as npimport pandas as pdimport scanpy as scimport seaborn as snsimport omicverse as ovimport matplotlib.pyplot as pltfrom matplotlib.pyplot import rc_context
import warningswarnings.filterwarnings('ignore')
plt.style.use('default')plt.rcParams['figure.facecolor'] = 'white'sc.settings.set_figure_params(dpi=100, dpi_save=200, figsize=(5, 5), facecolor='white')1. 8um bin数据读取
数据读取使用scanpy的read_visium函数即可,要注意一点,Visium HD使用的Space Ranger v3.0为了节省输出文件空间,将之前的spatial文件夹下的tissue_positions_list.csv文件格式变成了tissue_positions.parquet,为了使用read_visium函数正确读取数据,首先要将tissue_positions.parquet文件转成tissue_positions_list.csv文件。
data_dir = './CRC/P1_CRC/'tissue_position_df = pd.read_parquet(f'{data_dir}/binned_outputs/square_008um/spatial/tissue_positions.parquet')tissue_position_df.to_csv(f'{data_dir}/binned_outputs/square_008um/spatial/tissue_positions_list.csv', index=False, header=None)adata = sc.read_visium(f'{data_dir}/binned_outputs/square_008um', library_id='P1')adata.obs['sample'] = 'P1'adata.var_names_make_unique()2. 数据质控
空间转录组学实验中不可避免会引入一些技术噪音,比如由于实验处理或数据测序中的技术误差,导致部分细胞的测序数据异常(例如某些细胞可能由于实验处理不当或实际转录活性低,表现为转录本数量非常少)。细胞QC的目的是识别和过滤掉这些异常细胞,以避免它们对下游分析(如差异表达分析、聚类或功能分析)产生负面影响。通过去除低质量的细胞,我们可以确保保留下来的细胞是转录活性正常、具有代表性的细胞。这样不仅可以提高分析结果的生物学意义,还能减少假阳性结果。
# Data QCadata.var['mt'] = adata.var_names.str.upper().str.startswith('MT-') adata.var['ribo'] = adata.var_names.str.upper().str.startswith(("RPS","RPL"))sc.pp.calculate_qc_metrics(adata, qc_vars=['mt', 'ribo'], percent_top=None, log1p=False, inplace=True)adata.obs['log10GenesPerUMI'] = np.log10(adata.obs['n_genes_by_counts']) / np.log10(adata.obs['total_counts'])sc.pl.violin( adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'log10GenesPerUMI'], groupby='sample', jitter=False, rotation=90, multi_panel=True, show=False)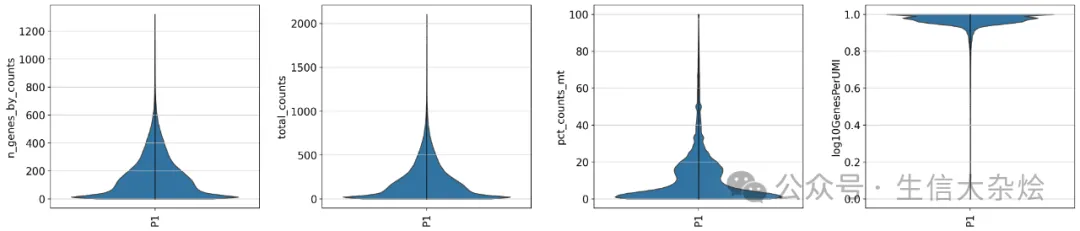
图片
3. 细胞过滤
低质量细胞会影响后续分析结果,这里我们和单细胞数据分析一样,首先将低质量细胞进行去除(当然也有些人不进行过滤,直接后续分析),这里我们将8um bin内鉴定到UMI counts数小于50个的细胞、线粒体基因占比大于30%的细胞、以及log10GenesPerUMI(主要反映的是细胞中鉴定到基因文库的复杂性)值小于0.8的细胞认为是低质量细胞,进行去除。
## Cell filtersc.pp.filter_cells(adata, min_counts=50)adata = adata[(adata.obs['pct_counts_mt'] < 30) & (adata.obs['log10GenesPerUMI'] > 0.8) & (adata.obs['log10GenesPerUMI'] < 0.99)]sc.pl.violin( adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'log10GenesPerUMI'], groupby='sample', jitter=False, rotation=90, multi_panel=True, show=False)
with rc_context({'figure.figsize': (8, 8)}): sc.pl.spatial( adata, color=['n_genes_by_counts', 'total_counts', 'log10GenesPerUMI'], library_id='P1', alpha_img=0.6 )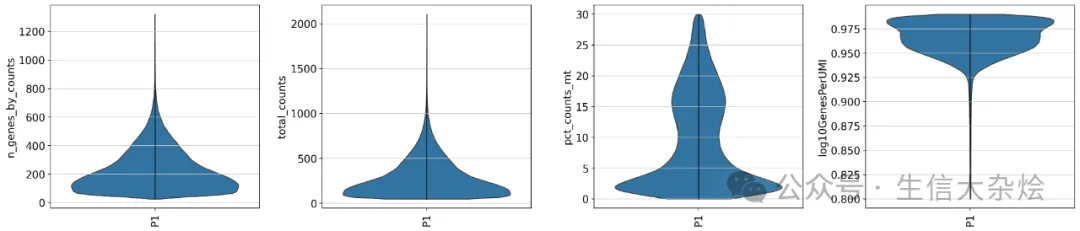
图片
细胞过滤后,空间上展示展示基因数、counts数等
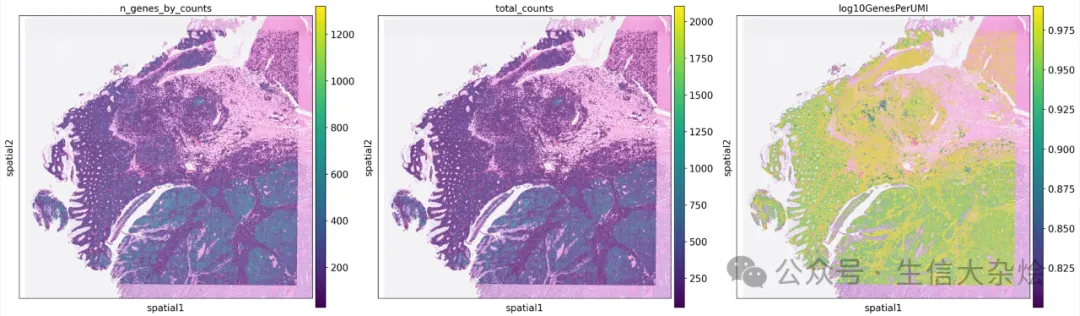
图片
4. 降维聚类
经过过滤之后的表达矩阵,使用Scanpy 进行降维聚类分析:
1)首先进行数据的表达量均一化,对测序深度进行校正,消除技术噪音;
2)选取高可变基因(细胞间高特征基因),用于提高细胞分群准确度(默认值为2000);
3)进一步对表达量值均一化处理后,利用PCA分析对数据进行降维,PCA降维是一种线性降维方法,运用方差分解,将高维的数据映射到低维的空间中;然后基于SNN聚类算法对细胞进行聚类和分群,构建细胞间的聚类关系;
4)最后将降维后的数据传递到UMAP进行可视化展示,细胞之间的基因表达模式越相似,在UMAP图中的距离也越接近。
## data normalization and log10 transformadata.layers["counts"] = adata.X.copy()sc.pp.normalize_total(adata)sc.pp.log1p(adata)adata.raw = adata ## select highly variable gene and data scale sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5, n_top_genes=2000)sc.pl.highly_variable_genes(adata, show=False)adata = adata[:, adata.var['highly_variable']]sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])sc.pp.scale(adata, max_value=10) ## PCAsc.tl.pca(adata, svd_solver='arpack')sc.pl.pca(adata, color='sample', show=False)sc.pl.pca_variance_ratio(adata, log=True, show=False) ## UMAPsc.pp.neighbors(adata, n_neighbors=15, n_pcs=15)sc.tl.umap(adata)sc.tl.leiden(adata, resolution=0.6, key_added='res_0.6')sc.tl.leiden(adata, resolution=0.8, key_added='res_0.8')降维聚类结果UMAP及空间展示
from matplotlib import patheffectsimport matplotlib.pyplot as pltfig, ax = plt.subplots(figsize=(6,6)) ov.pl.embedding(adata, basis='X_umap', color=['res_0.6'], size=4, show=False, legend_loc=None, add_outline=False, frameon='small', legend_fontoutline=2, ax=ax ) ov.utils.gen_mpl_labels( adata, 'res_0.6', exclude=("None",), basis='X_umap', ax=ax, adjust_kwargs=dict(arrowprops=dict(arrowstyle='-', color='black')), text_kwargs=dict(fontsize= 12 ,weight='bold', path_effects=[patheffects.withStroke(linewidth=2, foreground='w')] ),)单细胞降维聚类结果展示
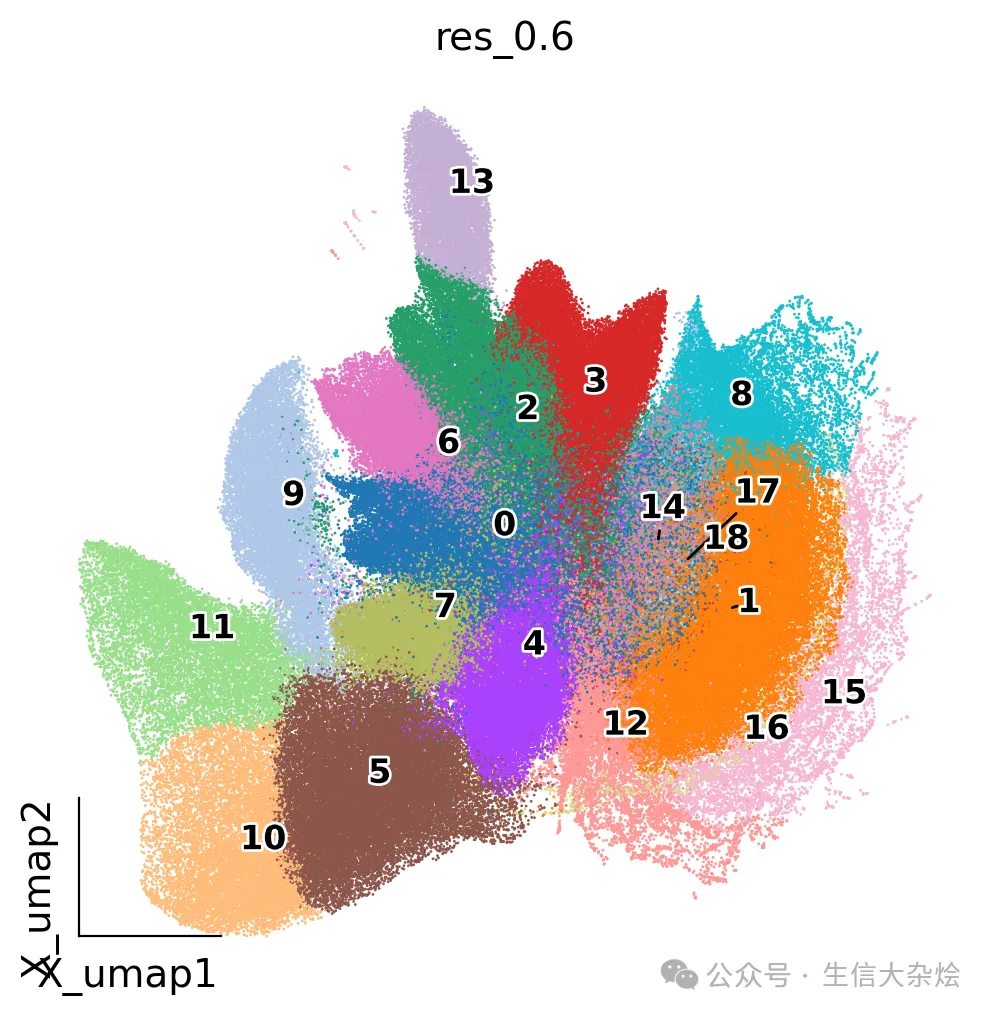
图片
降维聚类结果空间展示
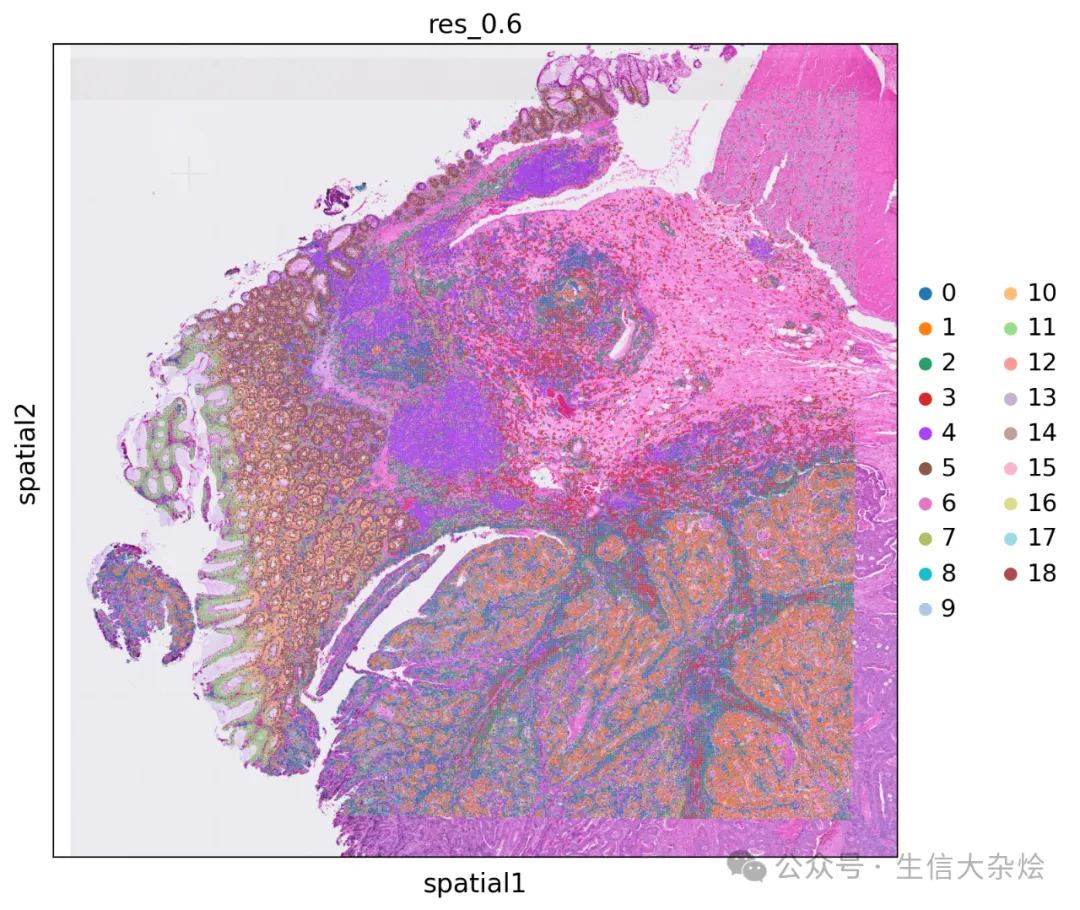
图片
特征基因筛选
sc.tl.rank_genes_groups( adata, groupby='res_0.6', use_raw=True, method="wilcoxon", pts=True) sc.pl.rank_genes_groups_dotplot( adata, groupby='res_0.6', standard_scale="var", n_genes=5, cmap='bwr', show=False,) result = adata.uns['rank_genes_groups']groups = result['names'].dtype.namesall_diff = []for c in groups: tmp = sc.get.rank_genes_groups_df(adata, group=c) tmp = tmp[(tmp['logfoldchanges']>0) & (tmp['pvals_adj']<0.05)] tmp['cluster'] = c all_diff.append(tmp) all_diff = pd.concat(all_diff, axis=0)各细胞Cluster Marker基因Dotplot, 结合上一步各Cluster特征基因,进行细胞类型定义。
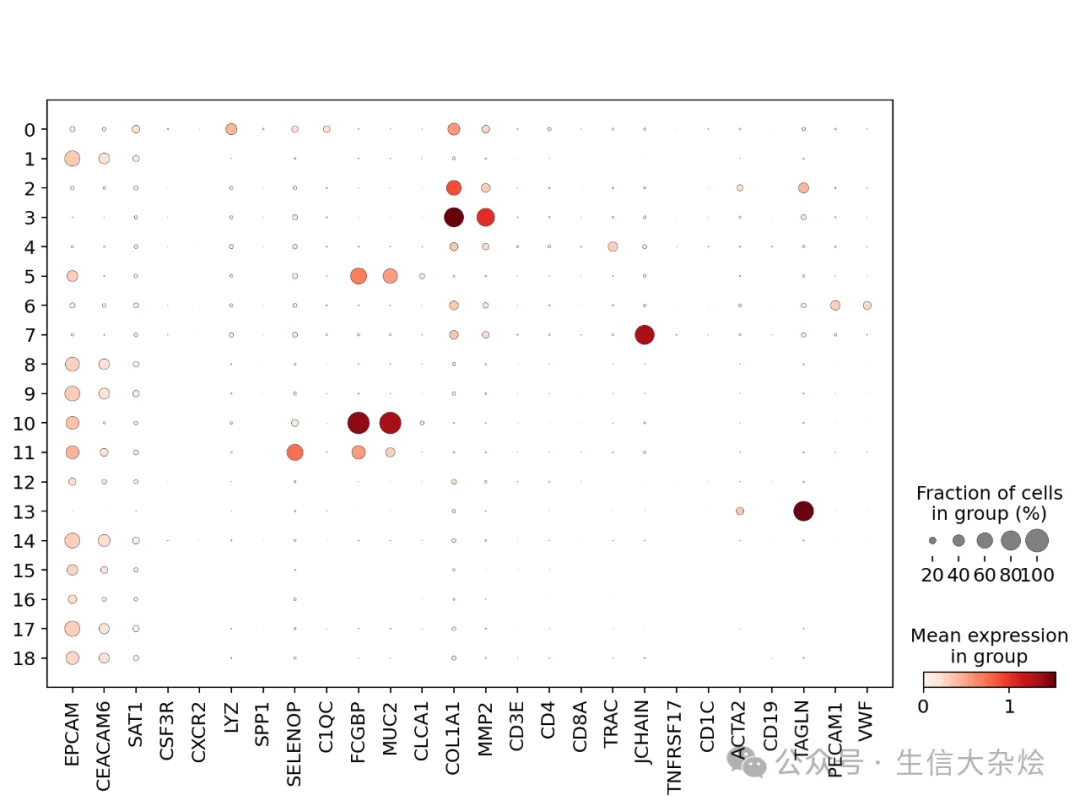
图片
这样我们就玩成了基础分析,接下来我们会介绍怎样进行细胞类型注释,以及为了获得单细胞精度的空间结果进行细胞核分割等分析。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号