GSVA和细胞通讯
原创

1 GSVA
什么是GSVA,可以用于分析基因集在不同样本或组中的表达差异情况。
1-3
#day 7
rm(list = ls())
library(Seurat)
rdaf = "sce.all.Rdata"
if(!file.exists(rdaf)){
f = dir("01_data/")
scelist = list() #创建空的列表,下面的for循环每执行一次,scelist里面就会多一个元素。
for(i in 1:length(f)){
pda <- Read10X(paste0("01_data/",f[[i]]))
scelist[[i]] <- CreateSeuratObject(counts = pda,
project = f[[i]],
min.cells = 3,
min.features = 200)
print(dim(scelist[[i]]))#输出每个文件的基因数和细胞数
}
sce.all = merge(scelist[[1]],scelist[-1]) #合并多个对象
sce.all = JoinLayers(sce.all)
#merge后,每个样本的表达矩阵是一个独立的的layer,JoinLayers是合并为一个表达矩阵
set.seed(335)
sce.all = subset(sce.all,downsample=700)#每个样本抽700个细胞,实战不允许
save(sce.all,file = rdaf)
}
load(rdaf)
head(sce.all@meta.data)
#质控
#添加指控指标需要的原素
sce.all[["percent.mt"]] <- PercentageFeatureSet(sce.all, pattern = "^MT-")
sce.all[["percent.rp"]] <- PercentageFeatureSet(sce.all, pattern = "^RP[SL]")
sce.all[["percent.hb"]] <- PercentageFeatureSet(sce.all, pattern = "^HB[^(P)]")
sce.all = subset(sce.all,percent.mt < 20&
nCount_RNA < 40000 &
nFeature_RNA < 6000)
table(sce.all@meta.data$orig.ident)
#整合降维聚类分群
f = "objday7.Rdata"
library(harmony)
if(!file.exists(f)){
sce.all = sce.all %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData(features = rownames(.)) %>%
RunPCA(pc.genes = VariableFeatures(.)) %>%
RunHarmony("orig.ident") %>%
FindNeighbors(dims = 1:15, reduction = "harmony") %>%
FindClusters(resolution = 0.5) %>%
RunUMAP(dims = 1:15,reduction = "harmony") %>%
RunTSNE(dims = 1:15,reduction = "harmony")
save(sce.all,file = f)
}
load(f)
scRNA = sce.all
#注释一下
library(celldex)
library(SingleR)
ls("package:celldex")
f = "single_ref/ref_BlueprintEncode.RData"
if(!file.exists(f)){
ref <- celldex::BlueprintEncodeData()
save(ref,file = f)
}
ref <- get(load(f))
library(BiocParallel)
scRNA = sce.all
test = scRNA@assays$RNA$data
pred.scRNA <- SingleR(test = test,
ref = ref,
labels = ref$label.main,
clusters = scRNA@active.ident)
pred.scRNA$pruned.labels
new.cluster.ids <- pred.scRNA$pruned.labels
names(new.cluster.ids) <- levels(scRNA)
scRNA <- RenameIdents(scRNA,new.cluster.ids)
save(scRNA,file = "scRNA.Rdata")
# 到这第7天的数据终于准备好啦,因为之前的都放在一起现在乱遭的,下次一定要注意这个问题。
#day10,单样本和多样本都可以
library(Seurat)
library(GSVA)
library(clusterProfiler)
#load("../../day7/scRNA.Rdata")
seu.obj = scRNA
table(Idents(seu.obj)) #为啥不一样呢,因为我没注释,好的回去注释一下
exp = AverageExpression(seu.obj)[[1]] # 平均值做GSVA分析
#exp = AggregateExpression(seu.obj)[[1]] #求和做伪bulk分析,
exp = as.matrix(exp)
exp = exp[rowSums(exp)>0,]
exp[1:4,1:4]
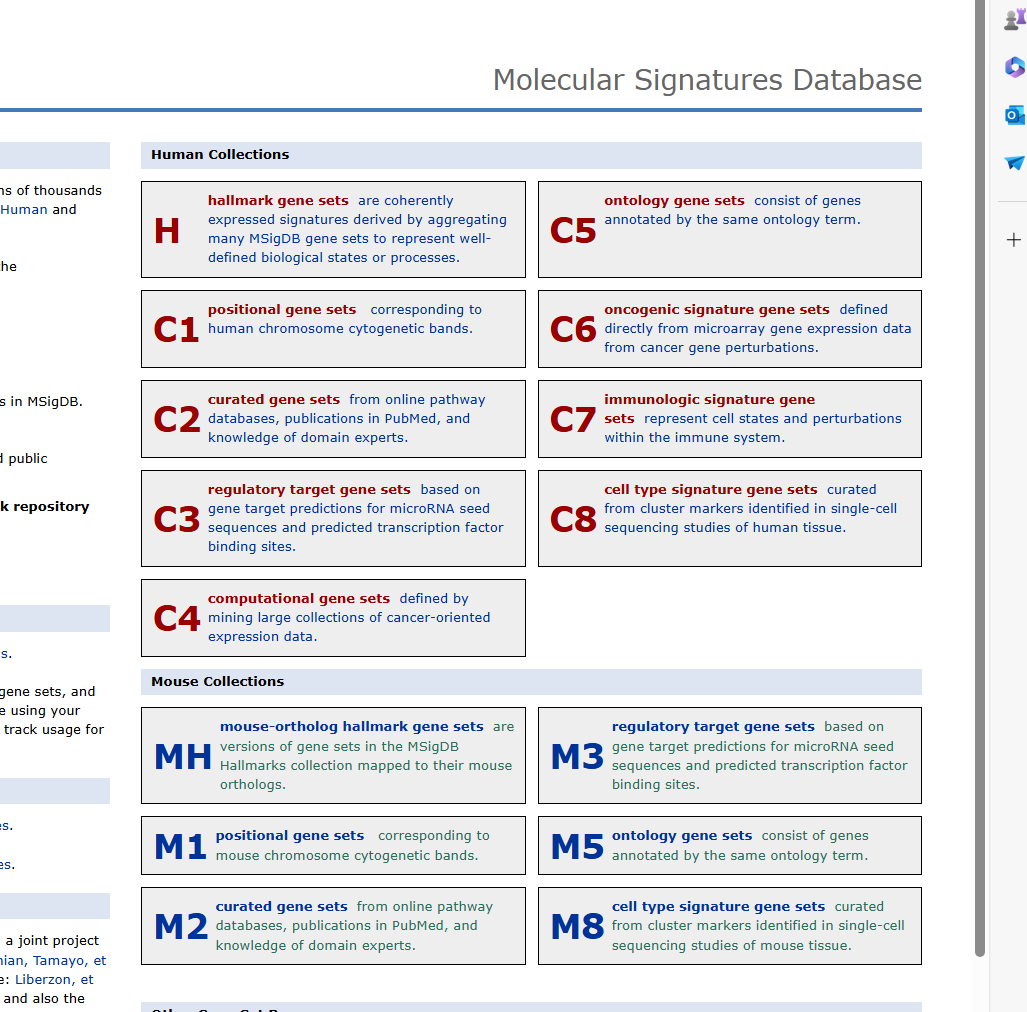
#https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp 下载gmt文件,no.1
h_df = read.gmt("day10/h.all.v2023.2.Hs.symbols.gmt")[,c(2,1)] #第二列和第一列交换位置了
h_list = unstack(h_df) #然后将这个交换后的矩阵转换为一个每个基因集对应一个列向量的数据结构,
?gsva #评价基因集的表达情况用zscoreParam
gsvapar <- zscoreParam(exp, h_list) #这个函数更新之后改写法了,和以前不太一样了
ES = gsva(gsvapar)
ES[1:4,1:4]
#热图可视化
library(pheatmap)
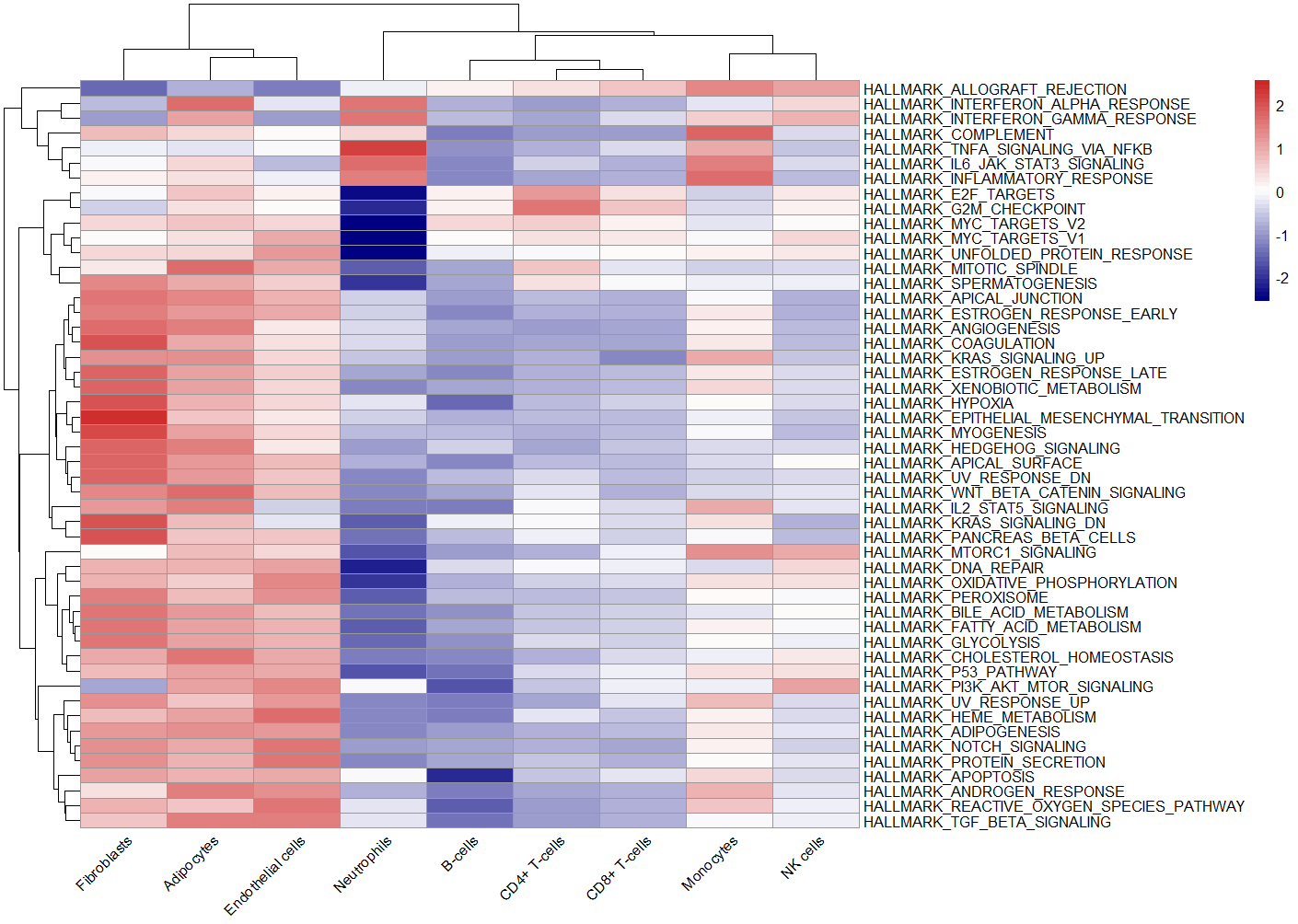
pheatmap(ES, scale = "row",angle_col = "45",
color = colorRampPalette(c("navy", "white", "firebrick3"))(50)) # no.2
no.1
no.2
1.4 多样本组间比较
上面的单样本多样本都可以,下面这一部分是多样本或多分组才u走·
#多样本
seu.obj$group = ifelse(seu.obj$orig.ident %in% paste0("sample",1:3),"treat","control")
exp = AverageExpression(seu.obj,group.by = c("ident","group"))[[1]]
#exp = AggregateExpression(seu.obj)[[1]]
exp = as.matrix(exp)
exp = exp[rowSums(exp)>0,]
exp[1:4,1:4]
#跟以前不太一样了,多了,"treat","control",因为这是两份组的
gsvapar <- zscoreParam(exp, h_list)
ES = gsva(gsvapar)
ES[1:4,1:4]
pheatmap(ES, scale = "row",angle_col = "45",
color = colorRampPalette(c("navy", "white", "firebrick3"))(50),
cluster_cols = F,
gaps_col = seq(2,ncol(ES)-1,2)) #no.3
pheatmap(ES, scale = "row",angle_col = "45",
color = colorRampPalette(c("navy", "white", "firebrick3"))(50),
cluster_cols = F) #no.4 去掉,gaps_col = seq(2,ncol(ES)-1,2)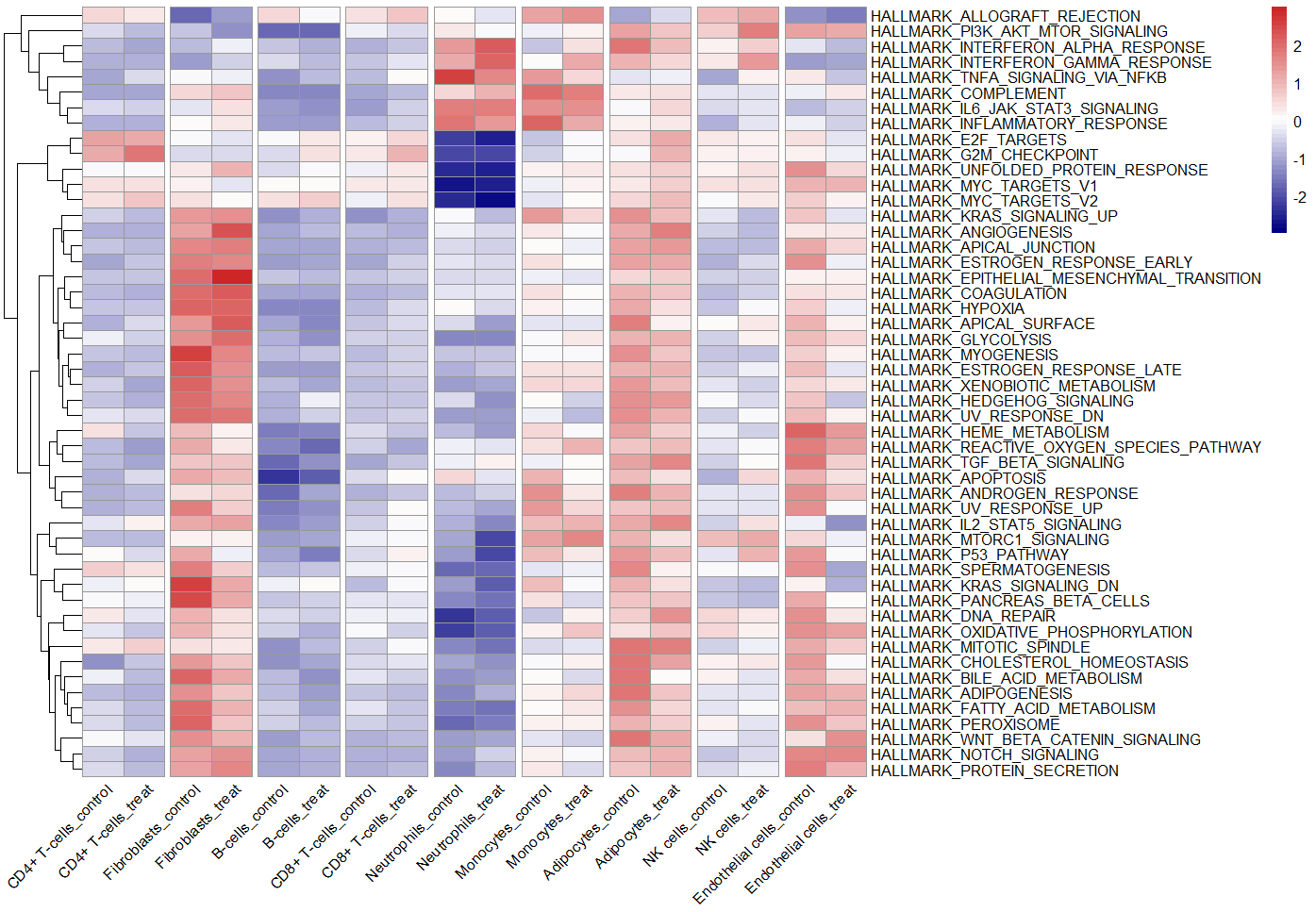
no.3组间热图
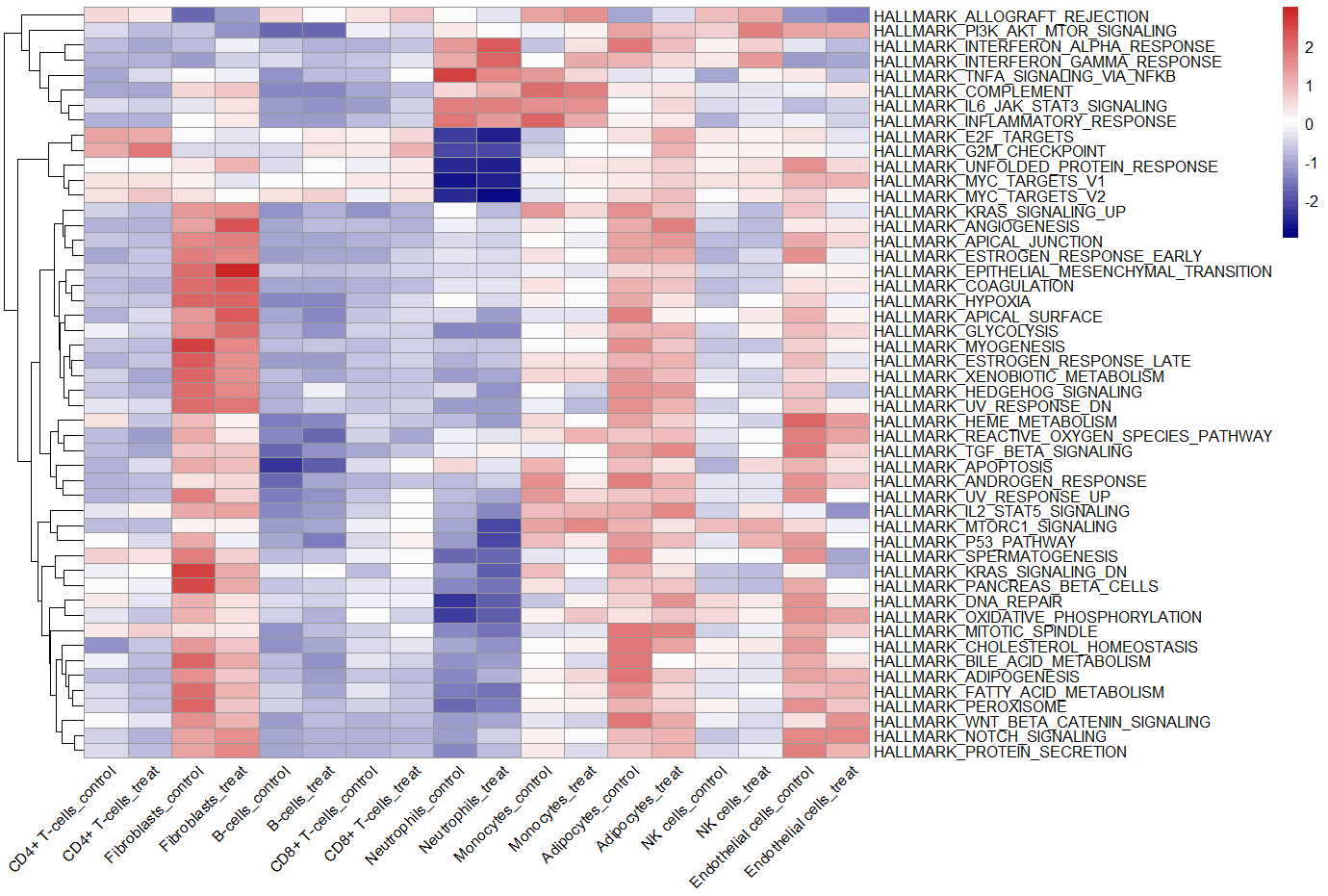
no.4 少了间隔
细胞通讯
细胞交流和响应外界刺激
教程:https://htmlpreview.github.io/?https://github.com/jinworks/CellChat/blob/master/tutorial/CellChat-vignette.html #再次感叹生信真牛
推荐教程:https://mp.weixin.qq.com/s/a2g25e3kc9EdfZcLcNgCJg 简单易懂
https://mp.weixin.qq.com/s/g4j7u0gHmpJH9ZA1Jp9hCw 用了点时间看
#细胞通讯用的day6的数据
Biocductor_packages <- c('sva',
'monocle',
'GOplot',
'GSVA',
'plotmo',
'regplot',
'scRNAseq',
'BiocStyle',
'celldex',
'SingleR',
'BiocParallel'
)
cran_packages <- c('tidyverse',
'msigdbr',
'patchwork',
'SeuratObject',
'Seurat'
)
for (pkg in c(Biocductor_packages,cran_packages)){
require(pkg,character.only=T) #require也可以换成library,livbrary没有error,require中一些warning需解决
}
library(Seurat)
library(patchwork)
library(tidyverse)
rm(list = ls())
library(Seurat)#加载这个Seurat这个包
load("obj0619.Rdata") #载入数据
p1 = DimPlot(seu.obj,reduction = "umap",label = T)
p1
library(dplyr)
f = "markers.Rdata"
if(!file.exists(f)){
markers <- FindAllMarkers(seu.obj, only.pos = TRUE,min.pct = 0.25)
save(markers,file = f)
}
load(f)
mks = markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)
g = unique(mks$gene)
library(ggplot2)
library(celldex)
library(SingleR)
ls("package:celldex")
z = "ref_Human_all.RData" #花花老师用的是f = "ref_BlueprintEncode.RData"
if(!file.exists(z)){
ref <- celldex::ref_Human_allData() #上面用啥这块改啥保留RData前面的内容,放在Data前面
save(ref,file = z)
}
ref <- get(load(z))
library(BiocParallel)
library(SingleR) #报错显示没找到,得加载一下
scRNA = seu.obj
test = scRNA@assays$RNA$data
pred.scRNA <- SingleR(test = test,
ref = ref,
labels = ref$label.main,
clusters = scRNA@active.ident)
pred.scRNA$pruned.labels #看一下分类
new.cluster.ids <- pred.scRNA$pruned.labels
names(new.cluster.ids) <- levels(scRNA)
library(Seurat) #"RenameIdents"是Seurat里面的
scRNA <- RenameIdents(scRNA,new.cluster.ids)
head(seu.obj@meta.data)
rm(list = ls())
options("repos"="https://mirrors.ustc.edu.cn/CRAN/")
if(!require("BiocManager")) install.packages("BiocManager",update = F,ask = F)
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
if(!require(devtools))install.packages("devtools")
if(!require(presto))devtools::install_github("immunogenomics/presto",upgrade = F,dependencies = T)
BiocManager::install("ComplexHeatmap")
if(!require(CellChat))devtools::install_local("CellChat-master/CellChat-master/",upgrade = F,dependencies = T) #注意看一下路径,要不很容易出问题
library(CellChat) #下载安装完在library
library(ggplot2)
library(Seurat)
library(ggalluvial)
#load("../../day6/sce.Rdata")
table(Idents(scRNA))
#scRNA = sce
DimPlot(scRNA)
#day10
rm(list = ls())
options("repos"="https://mirrors.ustc.edu.cn/CRAN/")
if(!require("BiocManager")) install.packages("BiocManager",update = F,ask = F)
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
if(!require(devtools))install.packages("devtools")
if(!require(presto))devtools::install_github("immunogenomics/presto",upgrade = F,dependencies = T)
BiocManager::install("ComplexHeatmap")
if(!require(CellChat))devtools::install_local("CellChat-master/CellChat-master/",upgrade = F,dependencies = T) #注意看一下路径,要不很容易出问题,报错说什么make的问题的话,重启一下R就行
library(CellChat) #下载安装完在library
library(ggplot2)
library(Seurat)
library(ggalluvial)
#load("../../day6/sce.Rdata")
load("scRNA.Rdata")
table(Idents(scRNA))
#scRNA = sce
DimPlot(scRNA) #no.1
scRNA = subset(scRNA,downsample = 100) #取子集,实际上不能这样做
table(Idents(scRNA)) #多余100的都变成了100个细胞
#CellChatDB.human,CellChatDB.mouse分别是人和小鼠的配受体数据库,根据物种按需修改
str(CellChatDB.human,max.level = 1)
table(CellChatDB.human$interaction$annotation)
#PPI.human和PPI.mouse是稀疏矩阵,是STRING数据库里高等级证据的相互作用关系组成的0-1矩阵,按需使用
class(PPI.human)
dim(PPI.human)
#table(as.numeric(PPI.human)) #费计算资源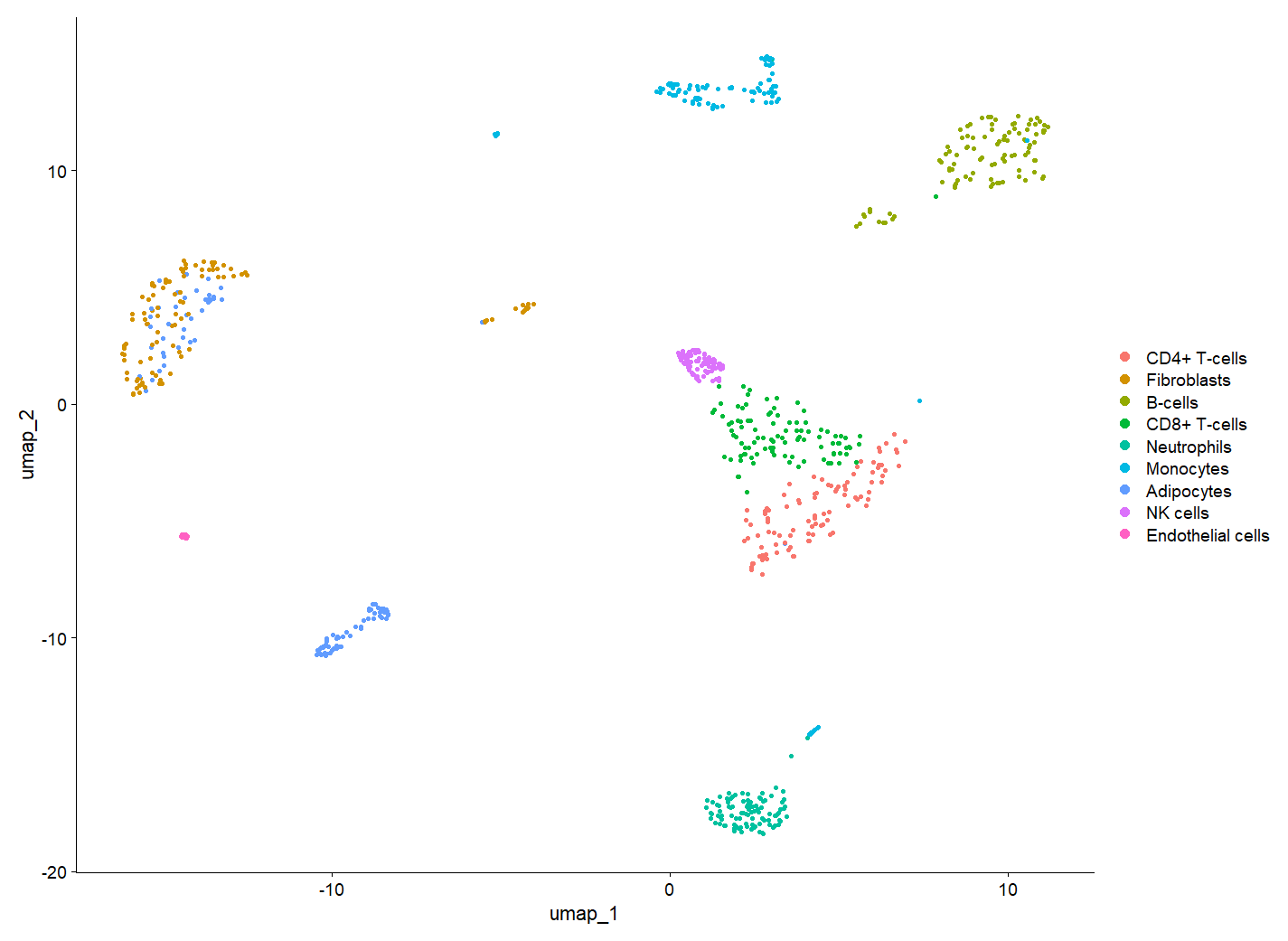
no.1
构建cellchat对象和细胞通讯网络分析及绘图展示
#构建cellchat对象
scRNA$samples = scRNA$orig.ident
cellchat <- createCellChat(scRNA,
group.by = "ident",
assay = "RNA")
cellchat@DB <- subsetDB(CellChatDB.human,
search = "Secreted Signaling")
#cellchat@DB可以按需修改,也可以用全部的cellchat@DB <- CellChatDB
cellchat <- subsetData(cellchat)
dim(cellchat@data.signaling)
#细胞通讯网络分析
cellchat <- identifyOverExpressedGenes(cellchat) # 识别过表达基因
# 识别配体-受体对
cellchat <- identifyOverExpressedInteractions(cellchat)
# 将配体、受体投射到PPI网络
cellchat <- projectData(cellchat, PPI.human)#慢,大约两分钟
# 推测细胞通讯网络
cellchat <- computeCommunProb(cellchat) #慢
cellchat <- computeCommunProbPathway(cellchat)
cellchat <- aggregateNet(cellchat)
#画图展示
cellchat@netP$pathways #分析出来的重要通路
pathways.show <- "GALECTIN" #根据上面出来的结果按需修改
#hierarchy plot
groupSize <- as.numeric(table(cellchat@idents))
vertex.receiver = seq(1,nlevels(scRNA)/2);vertex.receiver
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "hierarchy", vertex.receiver = vertex.receiver, vertex.weight = groupSize)
#circle plot #第二种展示方式,个人喜欢这种,比第一种好看
par(mfrow = c(1,1), xpd=TRUE,mar = c(2, 2, 2, 2))
netVisual_aggregate(cellchat, signaling = pathways.show,
layout = "circle",
vertex.receiver = vertex.receiver,
vertex.weight = groupSize) #no.2
#chord plot 第三种,这种也很好看,内圈小短线代表发射出的信号被谁接收,和接受者颜色一致,发射端是平的且有小短线,接收端是尖的
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "chord", vertex.receiver = vertex.receiver, vertex.weight = groupSize)
#heatmap #纵坐标是发射端,横坐标是接收端,有颜色代表横纵坐标所指的两类细胞之间有通讯,颜色深浅代表通讯概率。右侧和上方的条形图是该行/列通讯概率之和,这个图信息好多
netVisual_heatmap(cellchat, signaling = pathways.show, color.heatmap = "Reds")
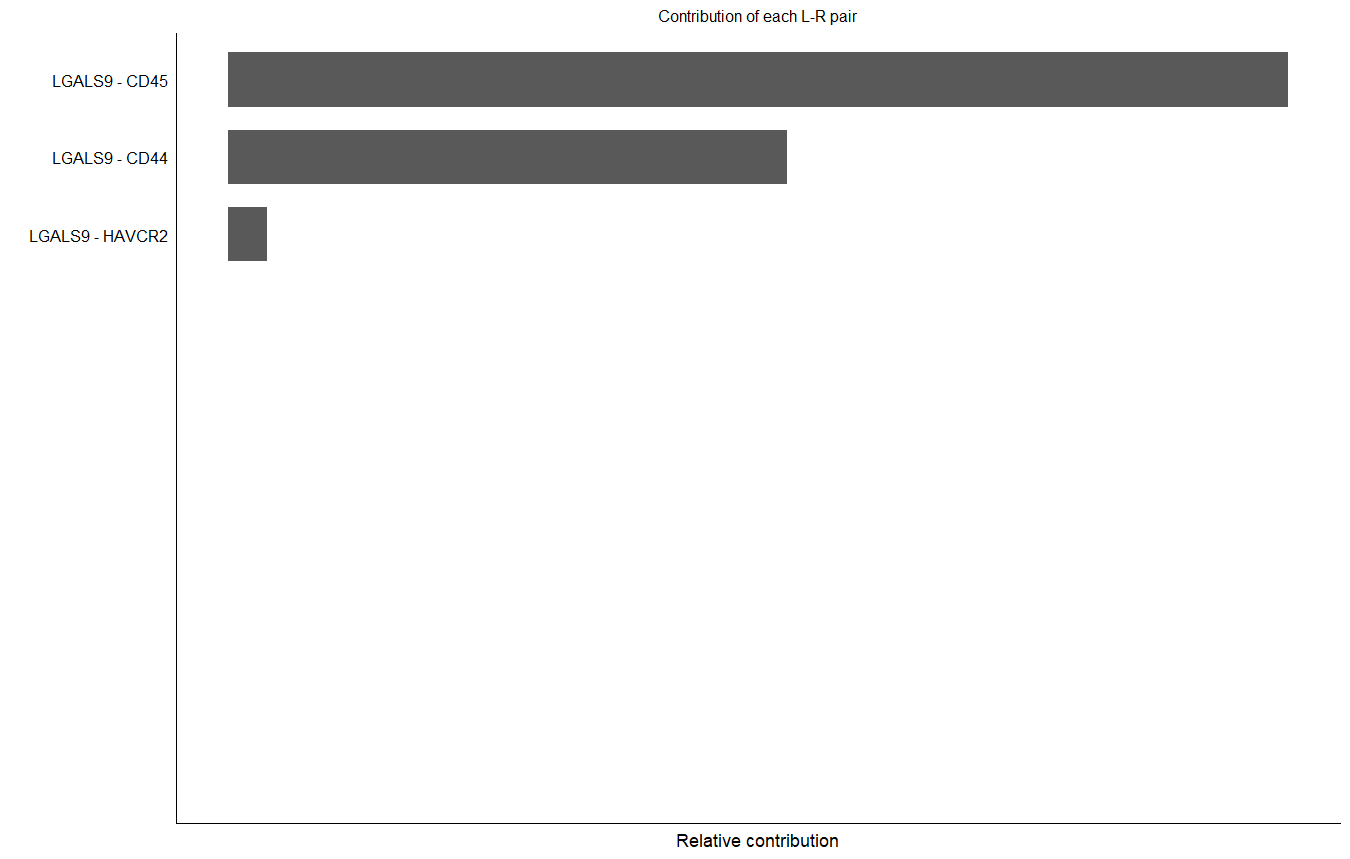
#计算配体-受体对信号网络的贡献度
netAnalysis_contribution(cellchat, signaling = pathways.show)
# 热图-展示每一类细胞是什么角色,发送者、接收者、调解者和影响者
cellchat <- netAnalysis_computeCentrality(cellchat, slot.name = "netP") # the slot 'netP' means the inferred intercellular communication network of signaling pathways
netAnalysis_signalingRole_network(cellchat, signaling = pathways.show, width = 12, height = 5, font.size = 10)
#气泡图-显示所有的显著的配体-受体对
#可以分开,也可以合到一起
netVisual_bubble(cellchat, sources.use = 1,
targets.use = 1:nlevels(scRNA),
remove.isolate = FALSE)
#从第一类细胞到全部细胞
netVisual_bubble(cellchat, sources.use = 1:nlevels(scRNA),
targets.use = 1:nlevels(scRNA),
remove.isolate = FALSE)#从全部细胞到全部细胞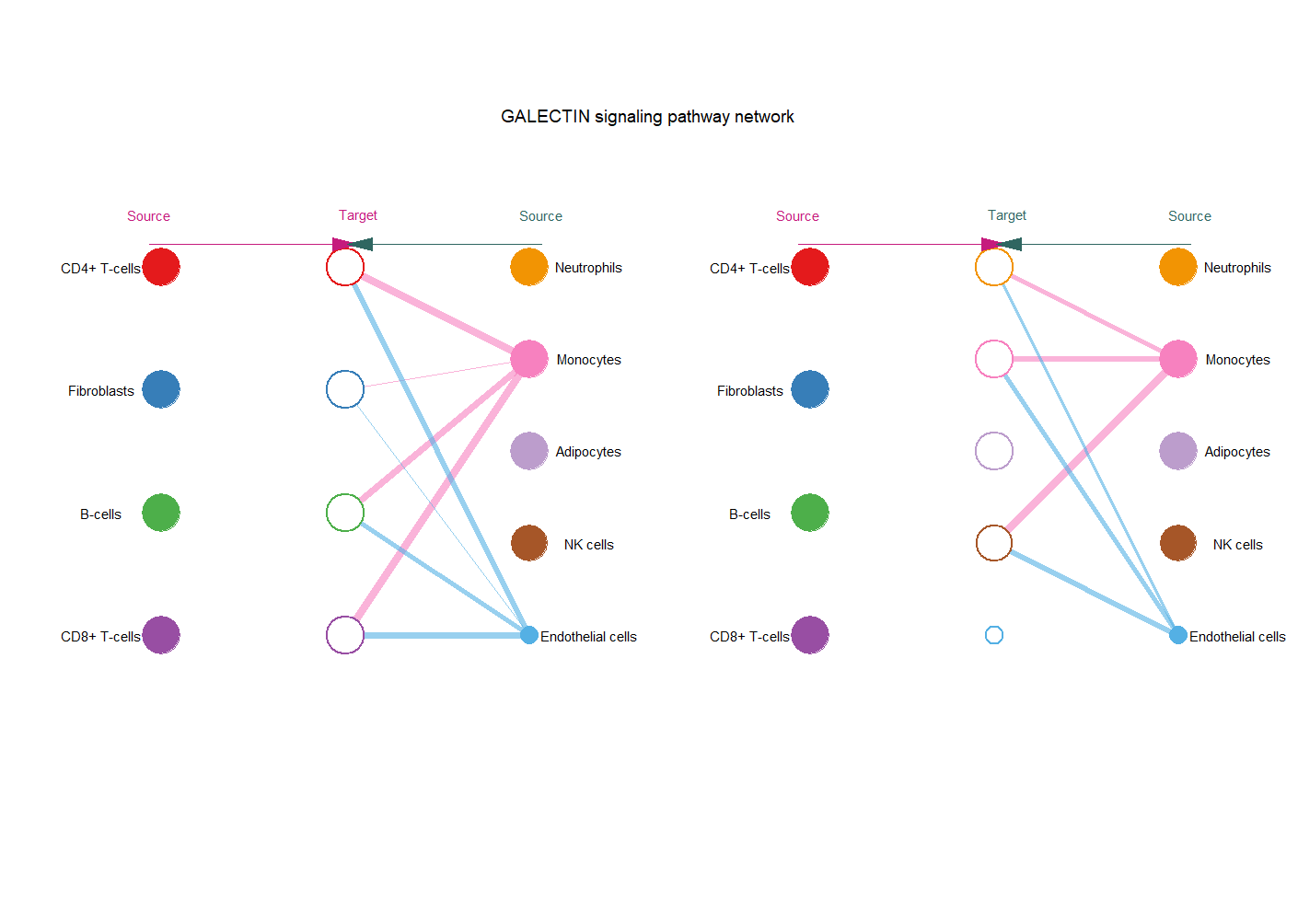
左图第二列等于第一列,右图第二列等于第三列
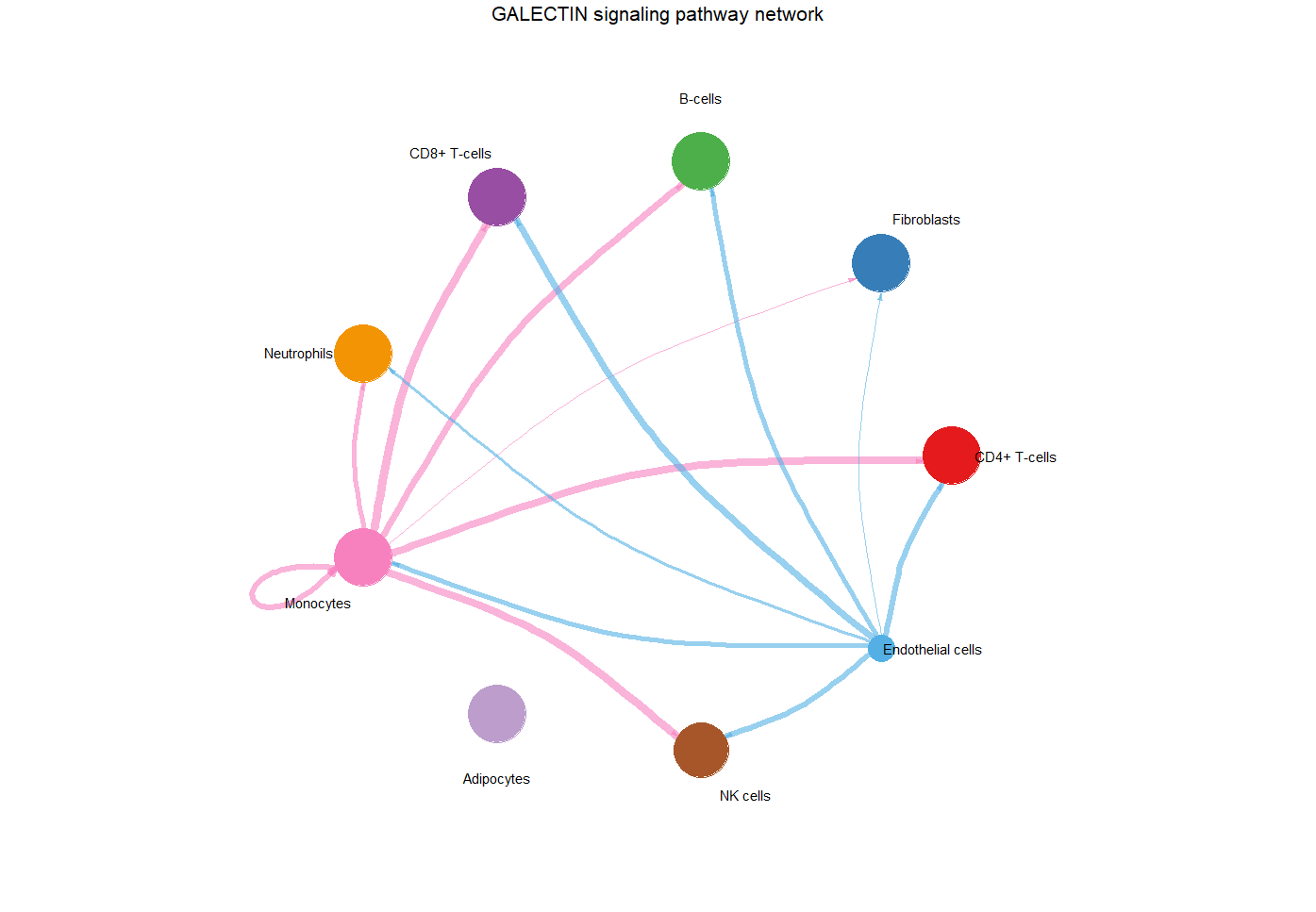
no.2circle plot
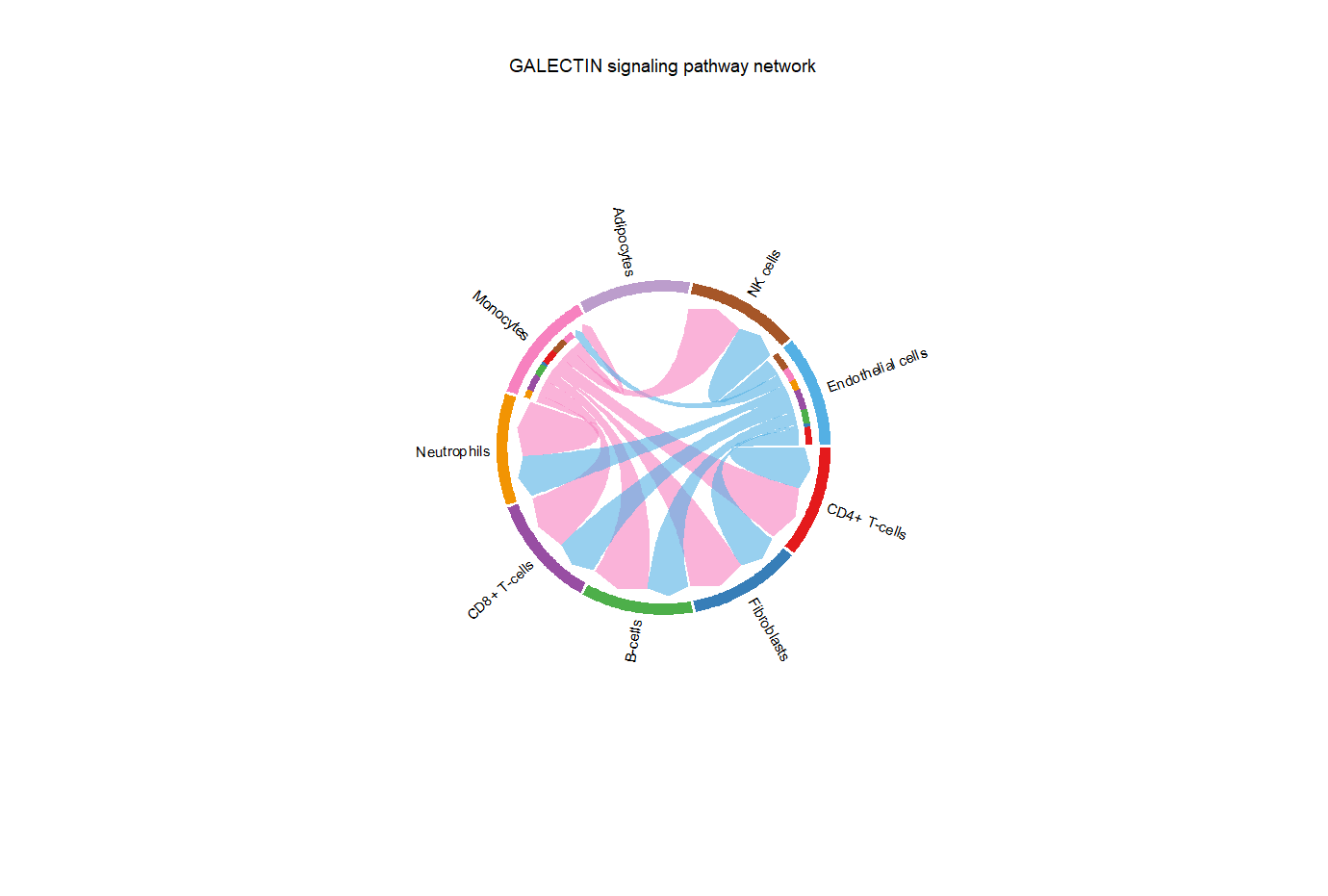
chord plot
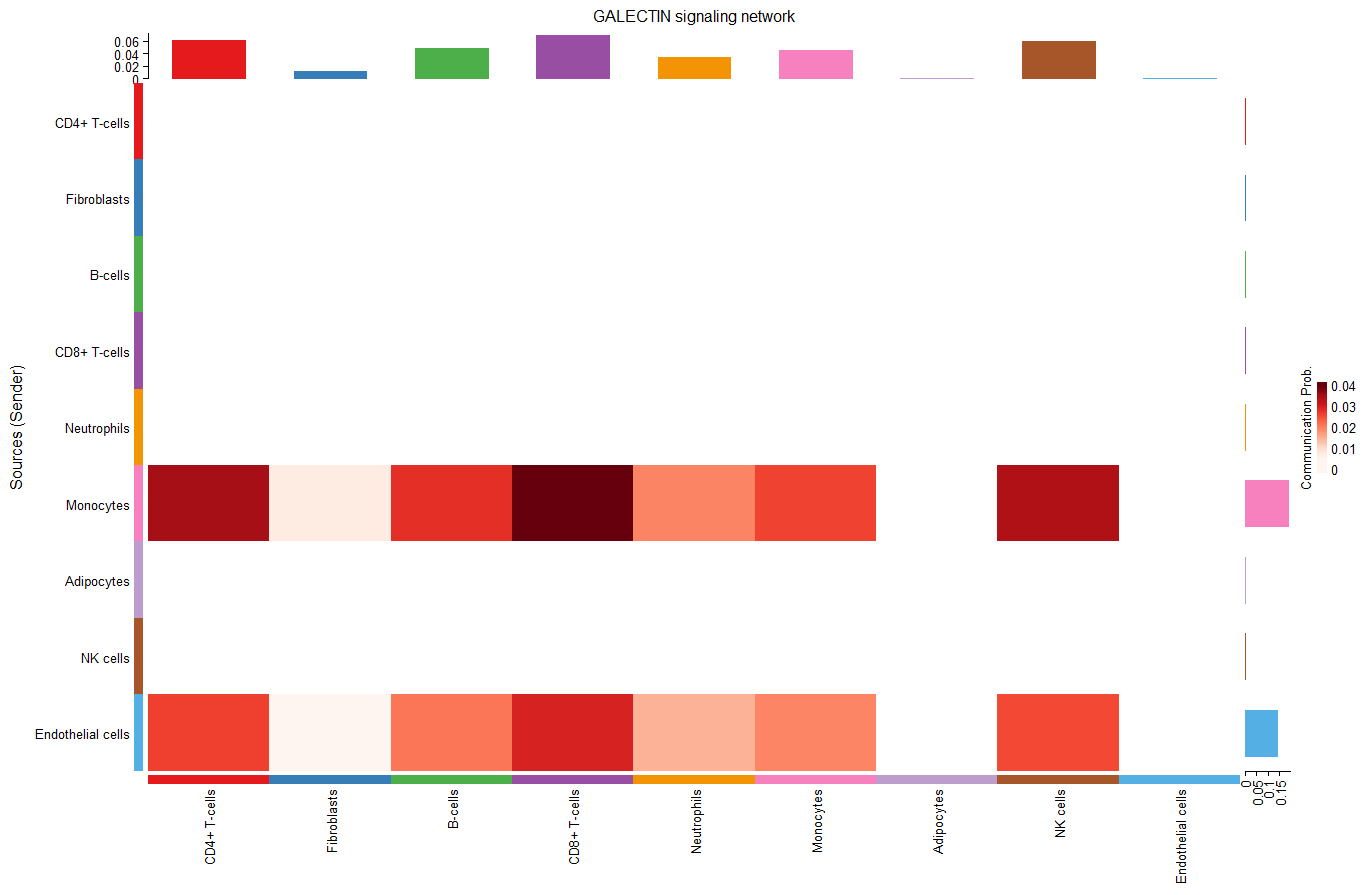
heatmap
纵坐标是发射端,横坐标是接收端,有颜色代表横纵坐标所指的两类细胞之间有通讯,颜色深浅代表通讯概率,上和右表示该列/行通讯概率之和
计算配体-受体对信号网络的贡献度
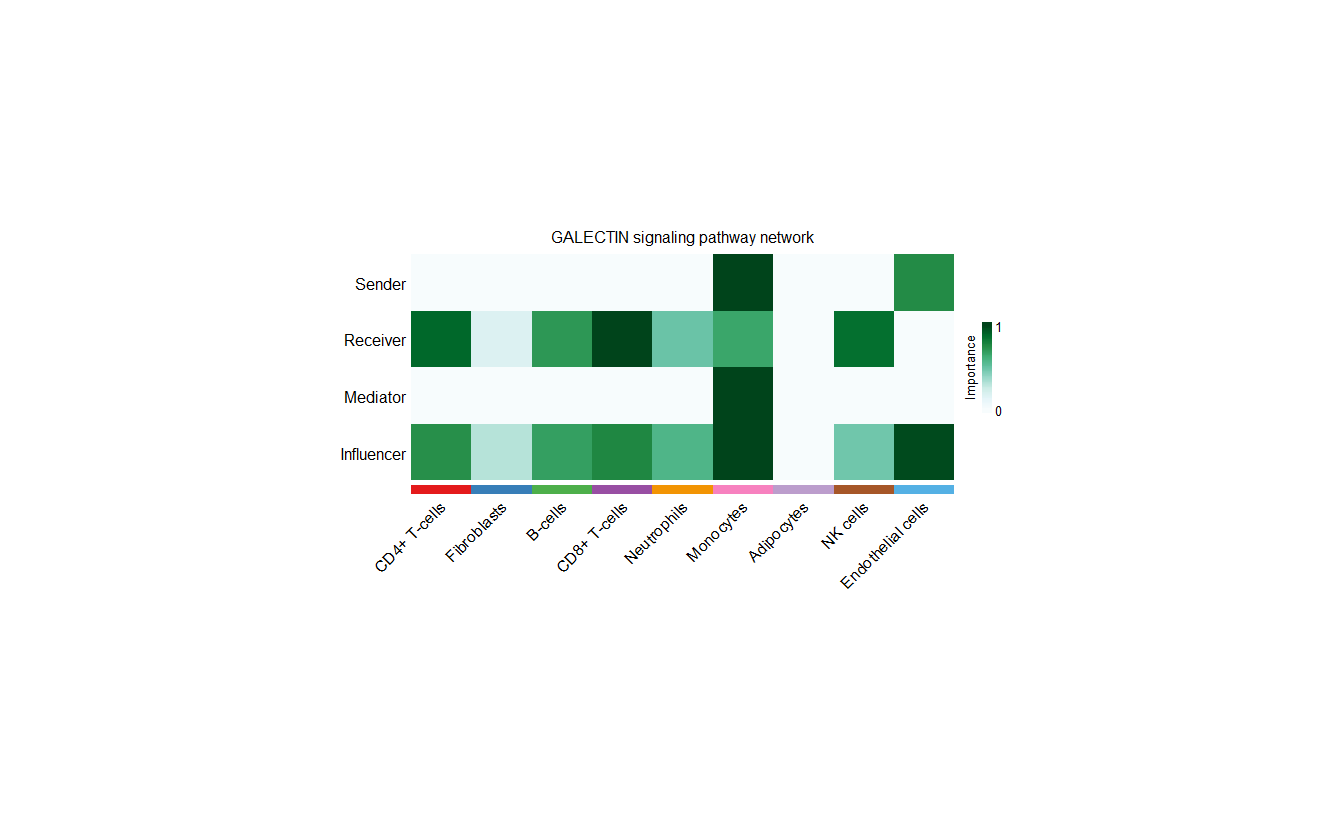
展示每一类细胞是什么角色,发送者、接收者、调解者和影响者
只有p<0.05的才会被画出来,颜色仍然是通讯概率,圈的大小是按照p值,p值越小圈越大。
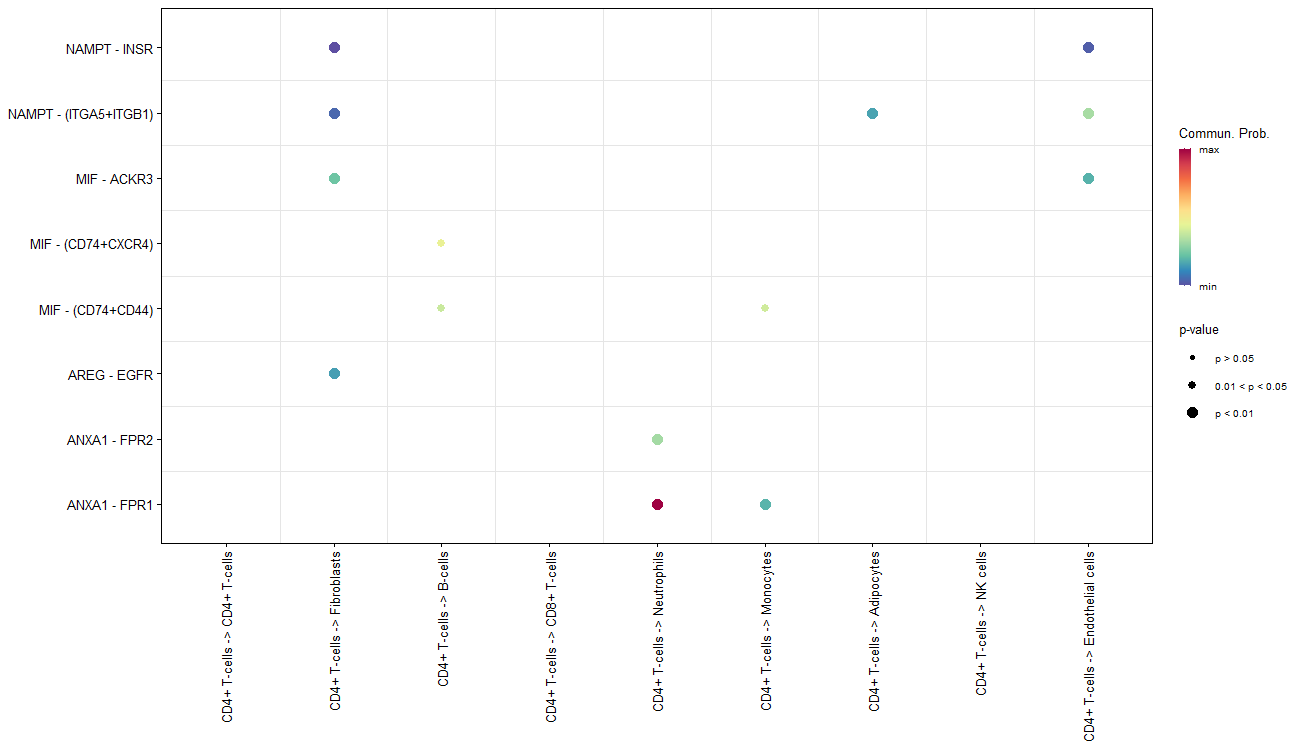
气泡图
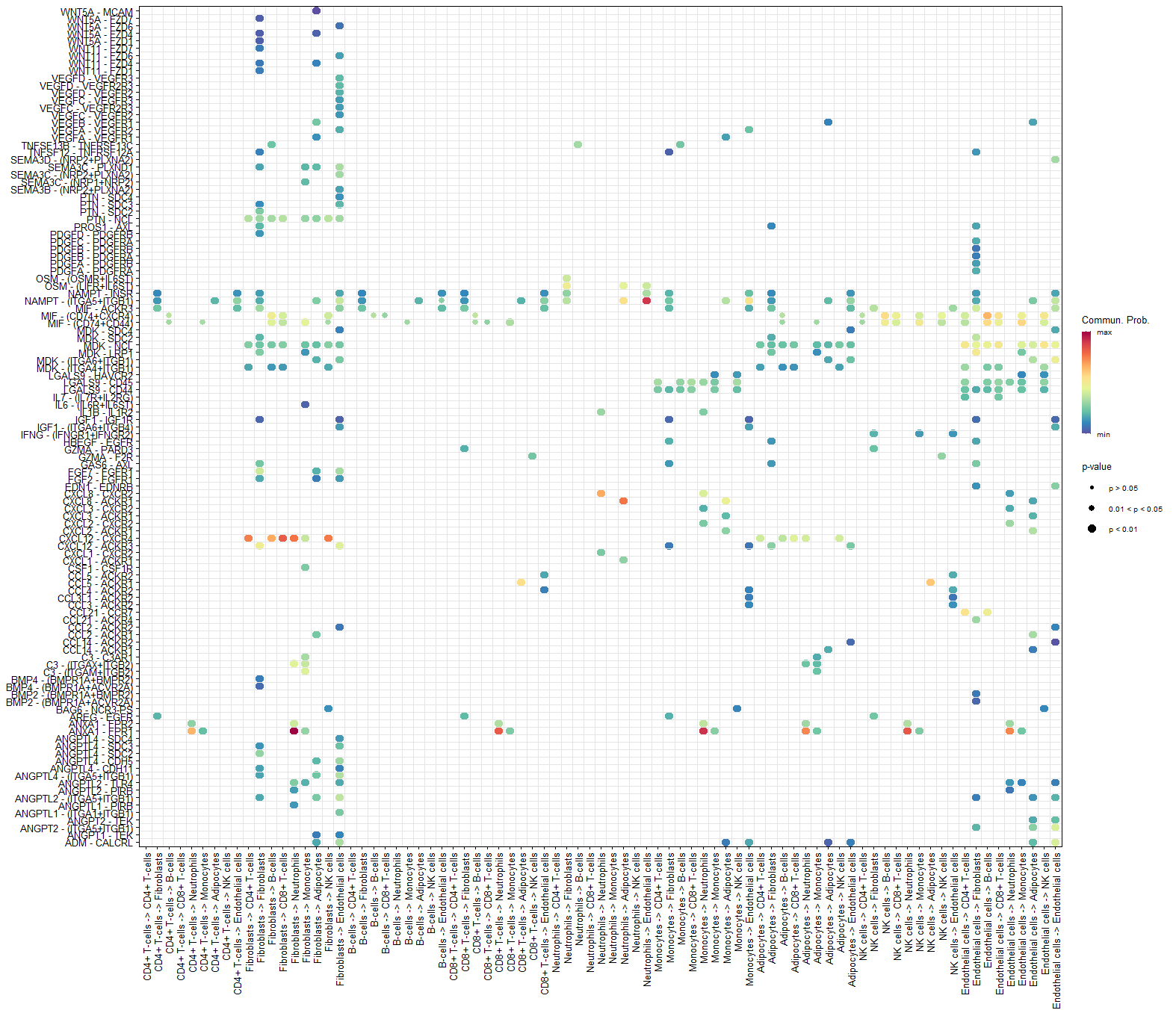
全部细胞到全部细胞
细胞通讯模式和信号网络
传出模式(outgoing),揭示了发射端细胞如何相互协调,以及它们如何与某些信号通路协调以驱动通信。
传入模式(incoming),显示接收端细胞如何相互协调,以及它们如何与某些信号通路协调以响应输入信号。
cellchat@netP$pathways #分析出来的重要通路
pathways.show <- "GALECTIN" #根据上面出来的结果按需修改
#hierarchy plot
groupSize <- as.numeric(table(cellchat@idents))
vertex.receiver = seq(1,nlevels(scRNA)/2);vertex.receiver
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "hierarchy", vertex.receiver = vertex.receiver, vertex.weight = groupSize)
#circle plot #第二种展示方式,个人喜欢这种,比第一种好看
par(mfrow = c(1,1), xpd=TRUE,mar = c(2, 2, 2, 2))
netVisual_aggregate(cellchat, signaling = pathways.show,
layout = "circle",
vertex.receiver = vertex.receiver,
vertex.weight = groupSize) #no.2
#chord plot 第三种,这种也很好看,内圈小短线代表发射出的信号被谁接收,和接受者颜色一致,发射端是平的且有小短线,接收端是尖的
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "chord", vertex.receiver = vertex.receiver, vertex.weight = groupSize)
#heatmap #纵坐标是发射端,横坐标是接收端,有颜色代表横纵坐标所指的两类细胞之间有通讯,颜色深浅代表通讯概率。右侧和上方的条形图是该行/列通讯概率之和,这个图信息好多
netVisual_heatmap(cellchat, signaling = pathways.show, color.heatmap = "Reds")
#计算配体-受体对信号网络的贡献度
netAnalysis_contribution(cellchat, signaling = pathways.show)
# 热图-展示每一类细胞是什么角色,发送者、接收者、调解者和影响者
cellchat <- netAnalysis_computeCentrality(cellchat, slot.name = "netP") # the slot 'netP' means the inferred intercellular communication network of signaling pathways
netAnalysis_signalingRole_network(cellchat, signaling = pathways.show, width = 12, height = 5, font.size = 10)
#气泡图-显示所有的显著的配体-受体对
#可以分开,也可以合到一起
netVisual_bubble(cellchat, sources.use = 1,
targets.use = 1:nlevels(scRNA),
remove.isolate = FALSE)
#从第一类细胞到全部细胞
netVisual_bubble(cellchat, sources.use = 1:nlevels(scRNA),
targets.use = 1:nlevels(scRNA),
remove.isolate = FALSE)#从全部细胞到全部细胞
# 细胞通讯模式和信号网络
library(NMF) #选择合适的nPatterns数,二者都突然下降的值对应的横坐标就是合适的聚类数,2到3突然下降就选2,3到4出现了下降就选3
selectK(cellchat, pattern = "outgoing")
nPatterns = 5
#根据上图选择的,嫌麻烦也可以用默认值5
#传出
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "outgoing", k = nPatterns)
selectK(cellchat, pattern = "incoming") #同理,输入也得来一遍这个过程
#nPatterns要根据上面的图来修改
#传入
nPatterns = 4
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "incoming", k = nPatterns)
#画图展示
#桑基图和气泡图展示每种细胞传入/传出的信号都是属于哪些通路的
# 桑基图
netAnalysis_river(cellchat, pattern = "outgoing")
netAnalysis_river(cellchat, pattern = "incoming")
# 气泡图
netAnalysis_dot(cellchat, pattern = "outgoing",dot.size = 4)
netAnalysis_dot(cellchat, pattern = "incoming",dot.size = 4)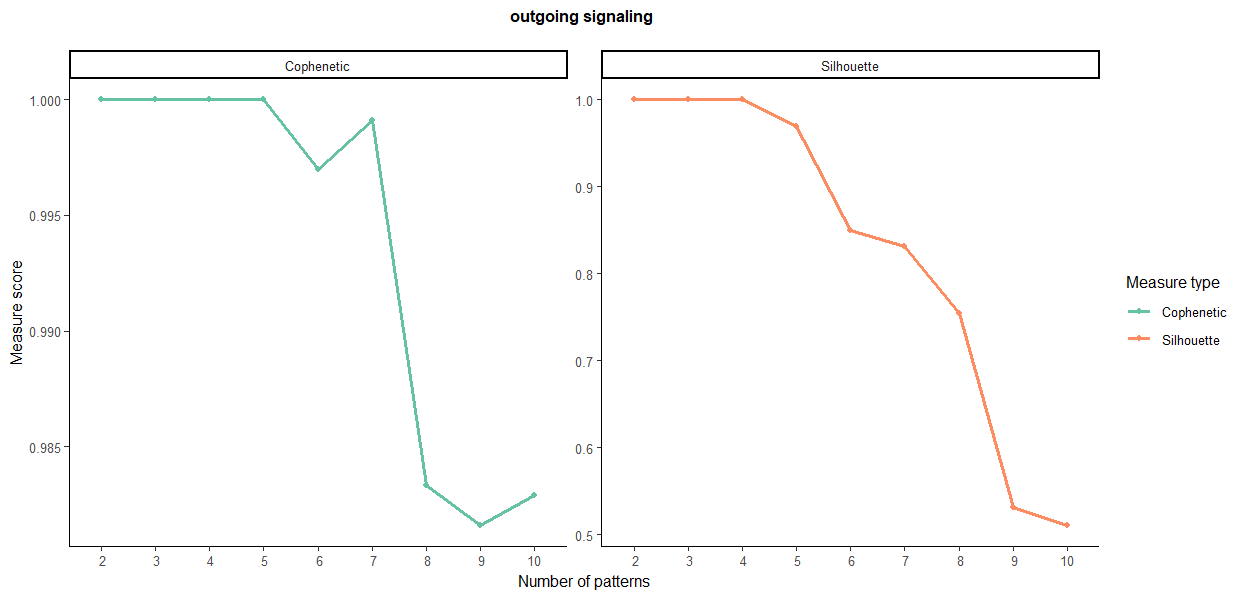
用的是patterns细胞,左图在5,右图在4,统一选5
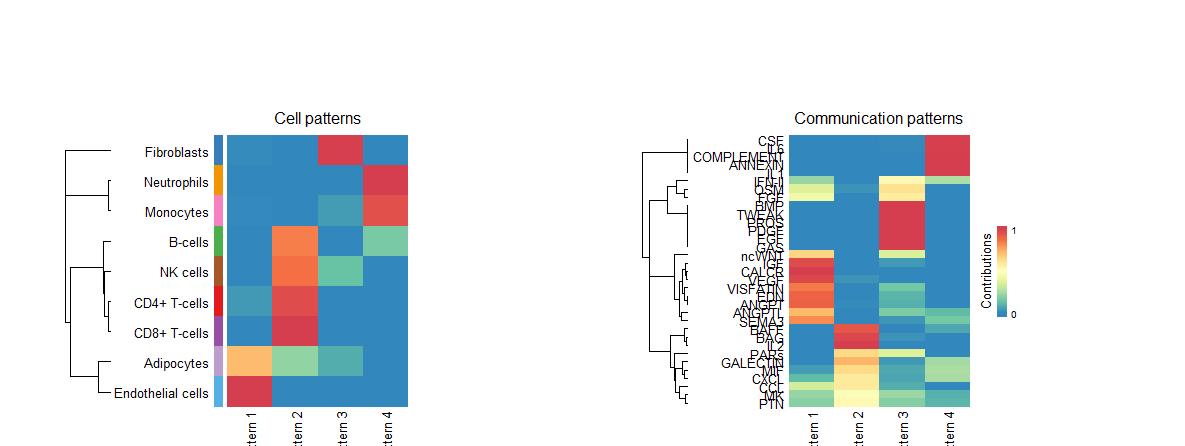
identifyCommunicationPatterns函数识别通讯模式,并画出热图
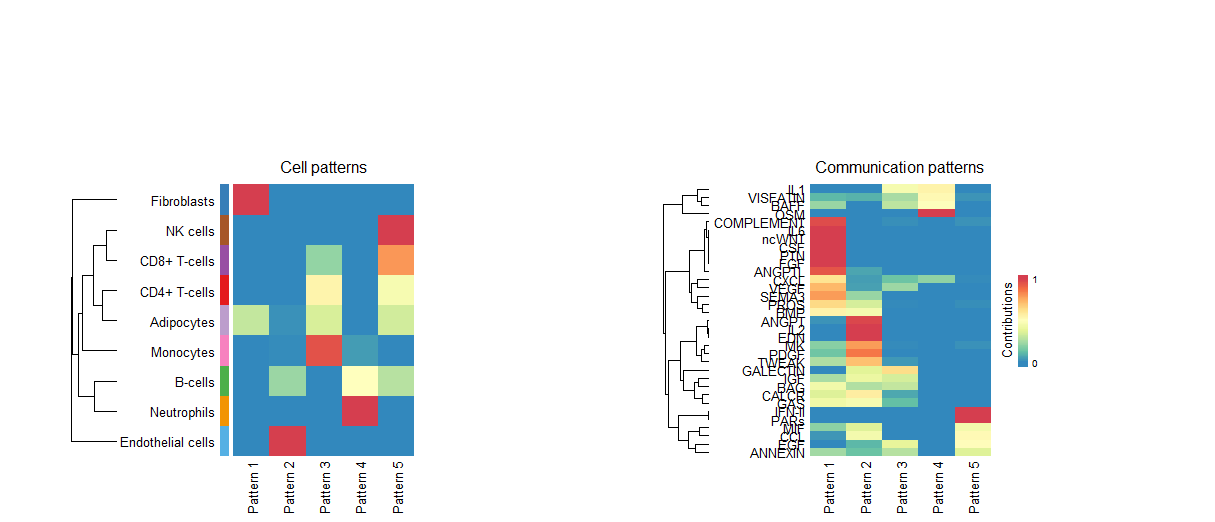
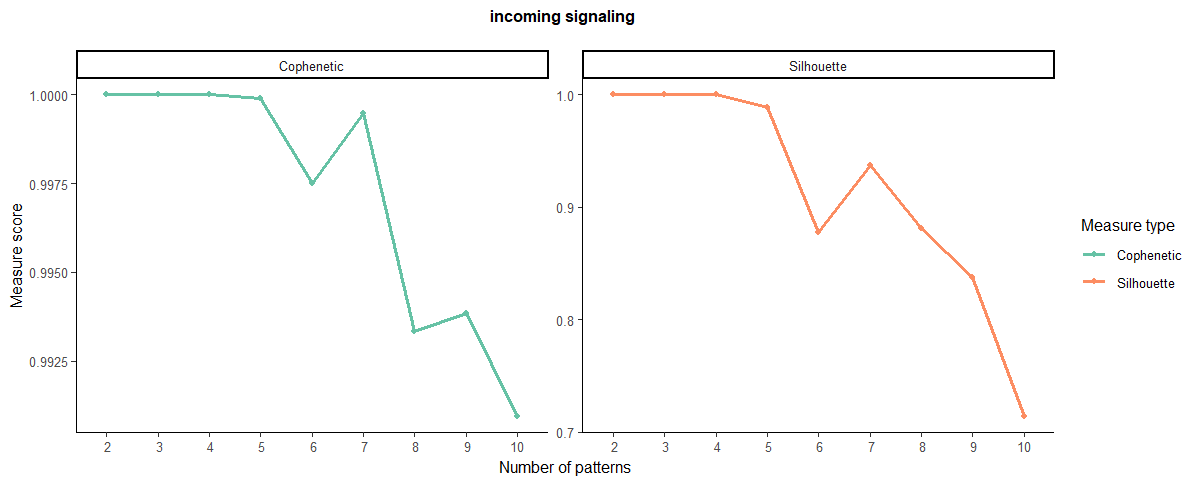
输入也得来一遍,选4
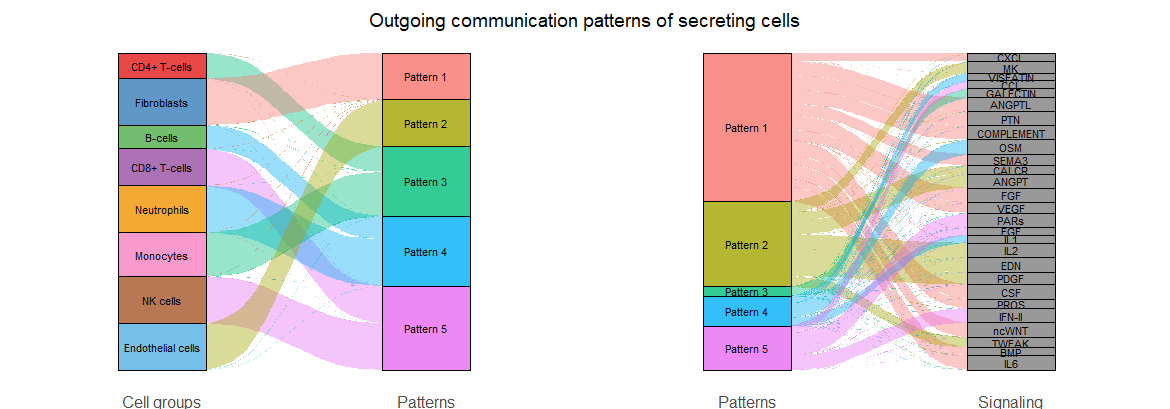
桑基图-输出
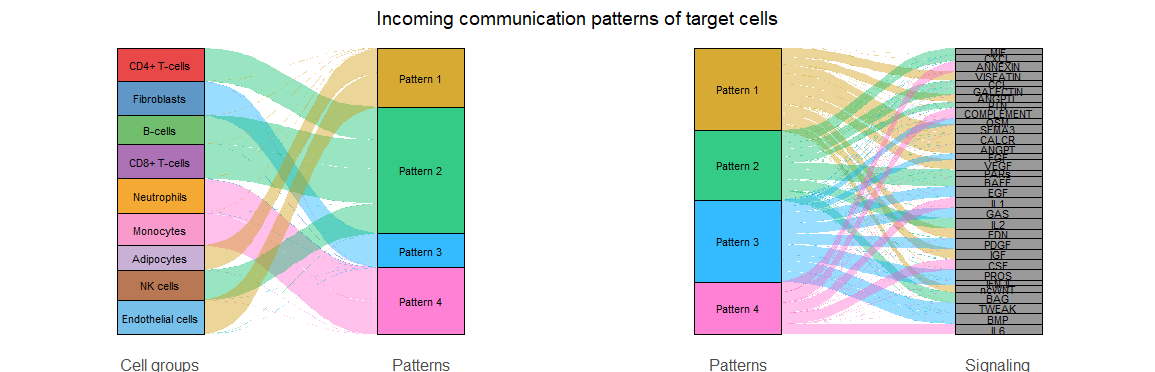
桑基图-输入
气泡图的颜色是按照细胞类型来分配,大小按照每个通路对每个细胞类型的贡献程度分配
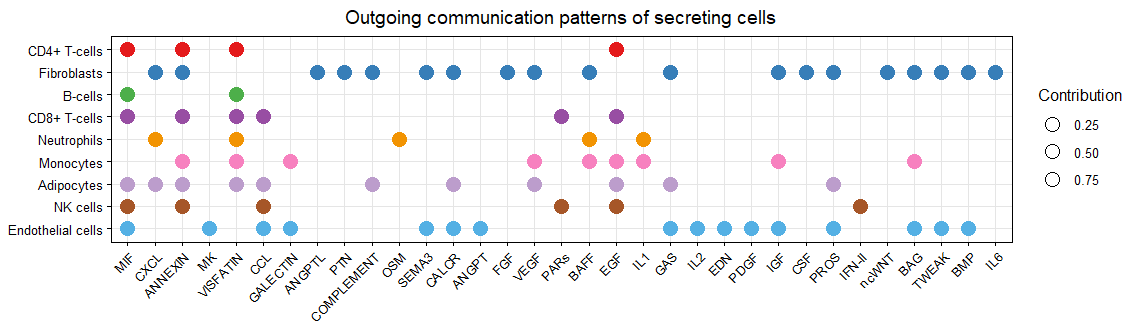
气泡图-输出
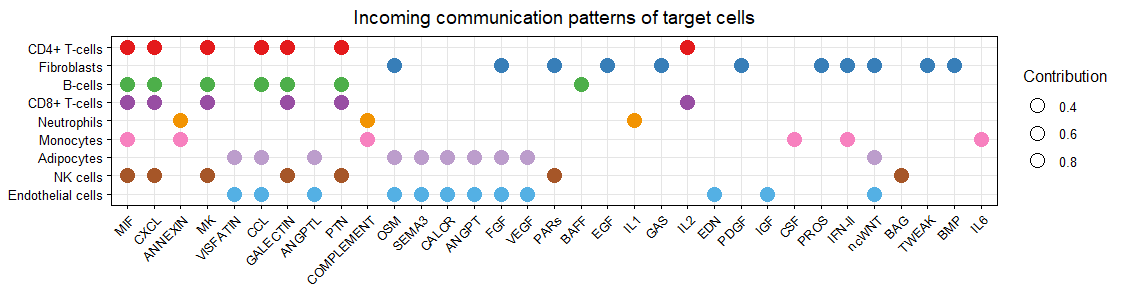
气泡图-输入
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号