单细胞学习小组003期 Day7
原创
单细胞学习小组003期 Day7
原创
用户11153857
发布于 2024-07-06 17:33:54
发布于 2024-07-06 17:33:54
Today, we do multiple samples of scRNA-seq.
Whole codes are from Huahua's tutorial and I collected them into one.
# 1. Download the dataset from GEO database, then save it at working directory
# https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi
untar("GSE231920_RAW.tar",exdir = "GSE231920_RAW") # unpack the file
library(stringr)
fs = paste0("GSE231920_RAW/",dir("GSE231920_RAW/"))
fs # file of data for each sample (fs)
samples = dir("GSE231920_RAW/") %>% str_split_i(pattern = "_",i = 2) %>% unique();samples # samples are the names of samples extracted from data files. The sample naming depends on the author, may be located in different loci of long name of file. Now, we know all names of samples are sample1, sample2, ..., and sample6.
# 2. Organise the data into 10X format
## 2.1 Create folders for each sample
# Here, lapply + self-defined function for bulk processing.
ctr = function(s){
ns = paste0("01_data/",s)
if(!file.exists(ns))dir.create(ns,recursive = T)
}
lapply(samples, ctr) # repeat using ctr(self-defined functiion), create folders for all samples under ./01_data
## 2.2 copy three files for each sample into the coresponding folders
lapply(fs, function(s){
#s = fs[1]
for(i in 1:length(samples)){
#i = 1
if(str_detect(s,samples[[i]])){
file.copy(s,paste0("01_data/",samples[[i]]))
}
}
})
# #s = fs[1] and #i = 1 in above codes are for testing when coding
## 2.3 Rename for all data in each folders
on = paste0("01_data/",dir("01_data/",recursive = T));on # define all names of all files by vector “on”
nn = str_remove(on,"GSM\\d+_sample\\d_");nn # design the new names
file.rename(on,nn) # rename
# 3. Read data
rm(list = ls())
library(Seurat)
rdaf = "sce.all.Rdata"
if(!file.exists(rdaf)){
f = dir("01_data/")
scelist = list() # create a blank list, for loop for one time,scelist will be added one new etem
for(i in 1:length(f)){
pda <- Read10X(paste0("01_data/",f[[i]]))
scelist[[i]] <- CreateSeuratObject(counts = pda,
project = f[[i]],
min.cells = 3,
min.features = 200)
print(dim(scelist[[i]])) # numbers of genes and cells for each sample
}
sce.all = merge(scelist[[1]],scelist[-1]) # merge all samples into one object of Seurat
sce.all = JoinLayers(sce.all) # After merge, each sample is an independent layer,then JoinLayers for combining them into one matrix.
set.seed(335) # this is for the numbers of randomised seeds
sce.all = subset(sce.all,downsample=700) # select 700 cells for each sample
save(sce.all,file = rdaf)
}
load(rdaf)
head(sce.all@meta.data)
table(sce.all$orig.ident) # show the numbers of each sample
sum(table(Idents(sce.all))) # total numbers of cells
# 4. Quality control
sce.all[["percent.mt"]] <- PercentageFeatureSet(sce.all, pattern = "^MT-")
sce.all[["percent.rp"]] <- PercentageFeatureSet(sce.all, pattern = "^RP[SL]")
sce.all[["percent.hb"]] <- PercentageFeatureSet(sce.all, pattern = "^HB[^(P)]")
head(sce.all@meta.data, 3)
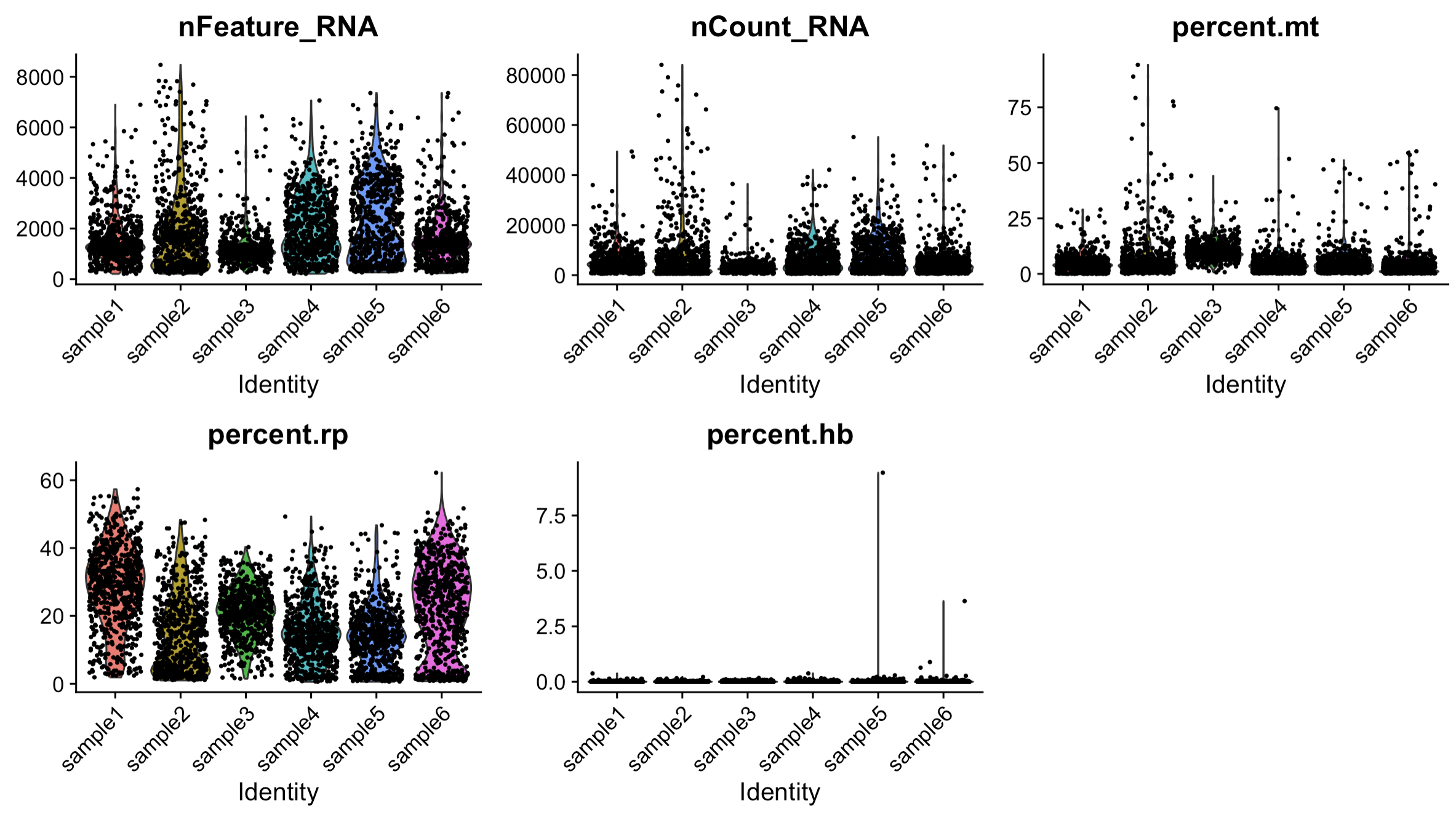
# Visualise the quality control results by Violin plot.
VlnPlot(sce.all,
features = c("nFeature_RNA",
"nCount_RNA",
"percent.mt",
"percent.rp",
"percent.hb"),
ncol = 3,pt.size = 0.5, group.by = "orig.ident")
# Based on the result of Violin plot, exclude the ouliers
sce.all = subset(sce.all,percent.mt < 20 &
nCount_RNA < 40000 &
nFeature_RNA < 6000)
table(sce.all@meta.data$orig.ident)
# 5. Integration of multiple samples by "harmony"
f = "obj.Rdata"
install.packages("harmony")
library(harmony)
if(!file.exists(f)){
sce.all = sce.all %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData(features = rownames(.)) %>%
RunPCA(pc.genes = VariableFeatures(.)) %>%
RunHarmony("orig.ident") %>%
FindNeighbors(dims = 1:15, reduction = "harmony") %>%
FindClusters(resolution = 0.5) %>%
RunUMAP(dims = 1:15,reduction = "harmony") %>%
RunTSNE(dims = 1:15,reduction = "harmony")
save(sce.all,file = f)
}
load(f)
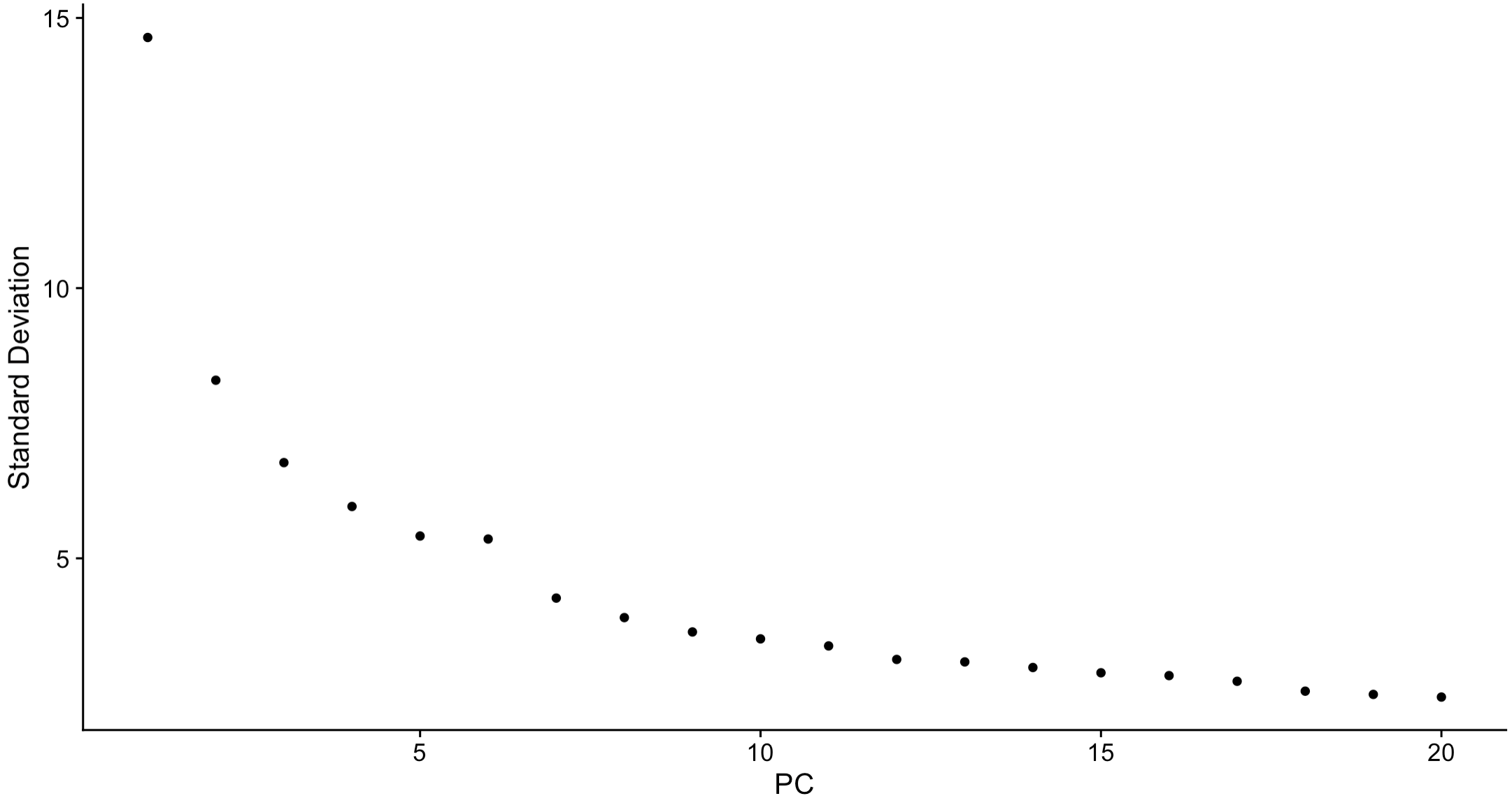
ElbowPlot(sce.all)
p1 = DimPlot(sce.all, reduction = "umap",label = T,pt.size = 0.5)+ NoLegend();p1
DimPlot(sce.all, reduction = "umap",pt.size = 0.5,group.by = "orig.ident")
# 6. Cell type annotation
# Cell type annotation for multiple samples is the same as for single sample.
library(celldex)
library(SingleR)
ls("package:celldex")
library(celldex)
library(SingleR)
ls("package:celldex")
f = "../Day6/ref_BlueprintEncode.RData" # employ the data of day6
if(!file.exists(f)){
ref <- celldex::BlueprintEncodeData()
save(ref,file = f)
}
ref <- get(load(f))
library(BiocParallel)
scRNA = sce.all
test = scRNA@assays$RNA$data
pred.scRNA <- SingleR(test = test,
ref = ref,
labels = ref$label.main,
clusters = scRNA@active.ident)
pred.scRNA$pruned.labels
new.cluster.ids <- pred.scRNA$pruned.labels
names(new.cluster.ids) <- levels(scRNA)
scRNA <- RenameIdents(scRNA,new.cluster.ids)
save(scRNA,file = "scRNA.Rdata")
p2 <- DimPlot(scRNA, reduction = "umap",label = T,pt.size = 0.5) + NoLegend()
p1+p2
# 7. Visualisation of grouping and comparisons of cell composition between groups
scRNA$seurat_annotations = Idents(scRNA) # add a column "meta.data" for meta information
## 7.1 Check the grouping information in GSE website or download from database and extract by yourself.
install.packages("tinyarray")
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clusterProfiler")
install.packages("Biobase")
install.packages('AnnoProbe')
BiocManager::install("GEOquery")
library(tinyarray)
edat = geo_download("GSE231920")
pd = edat$pd
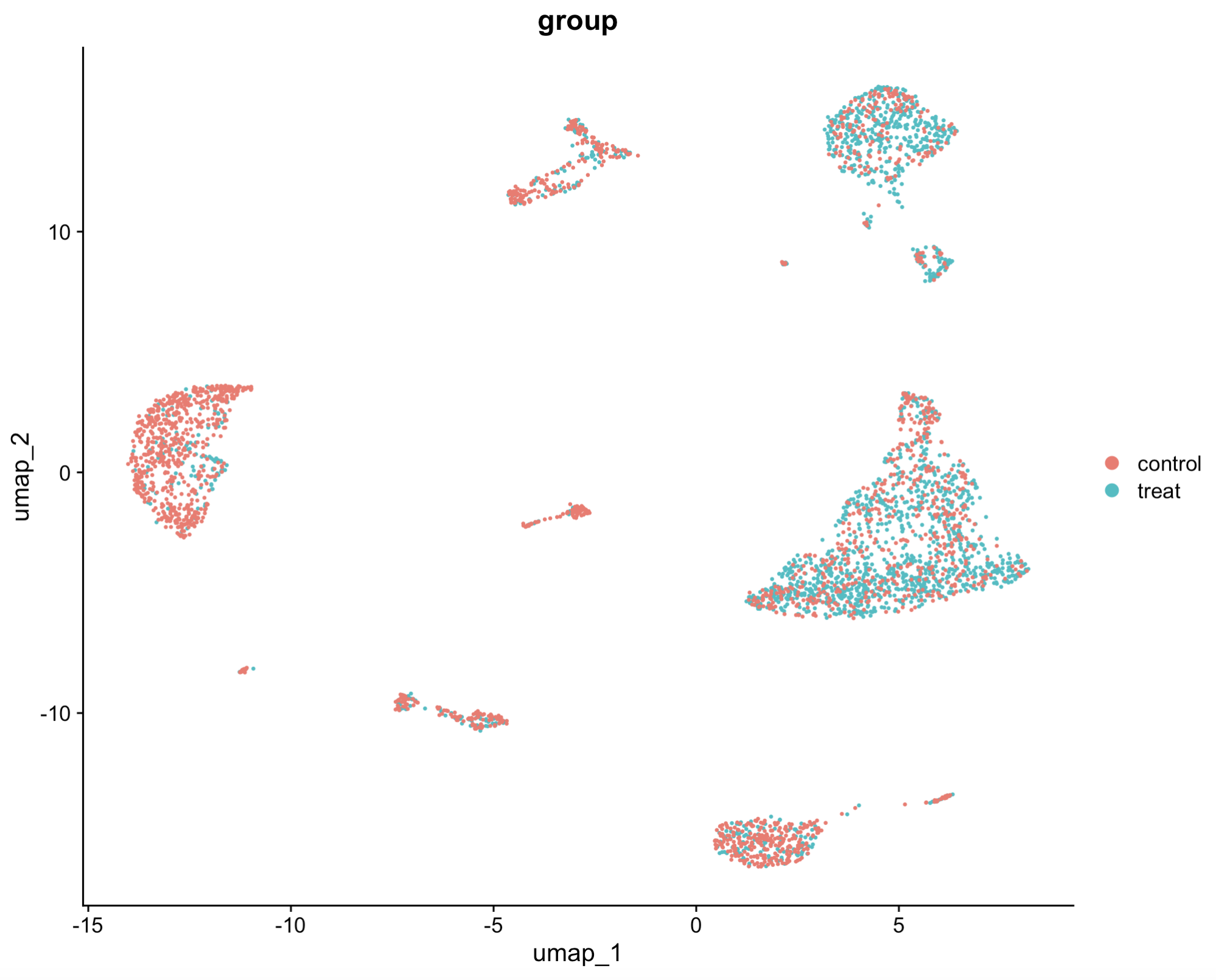
## 7.2 Add the group information as a new column, note them on DimPlot
scRNA$group = ifelse(scRNA$orig.ident %in% c("sample1","sample2","sample3"), "treat","control")
DimPlot(scRNA, reduction = "umap", group.by = "group") # scRNA$group works equally as scRNA@meta.data$group
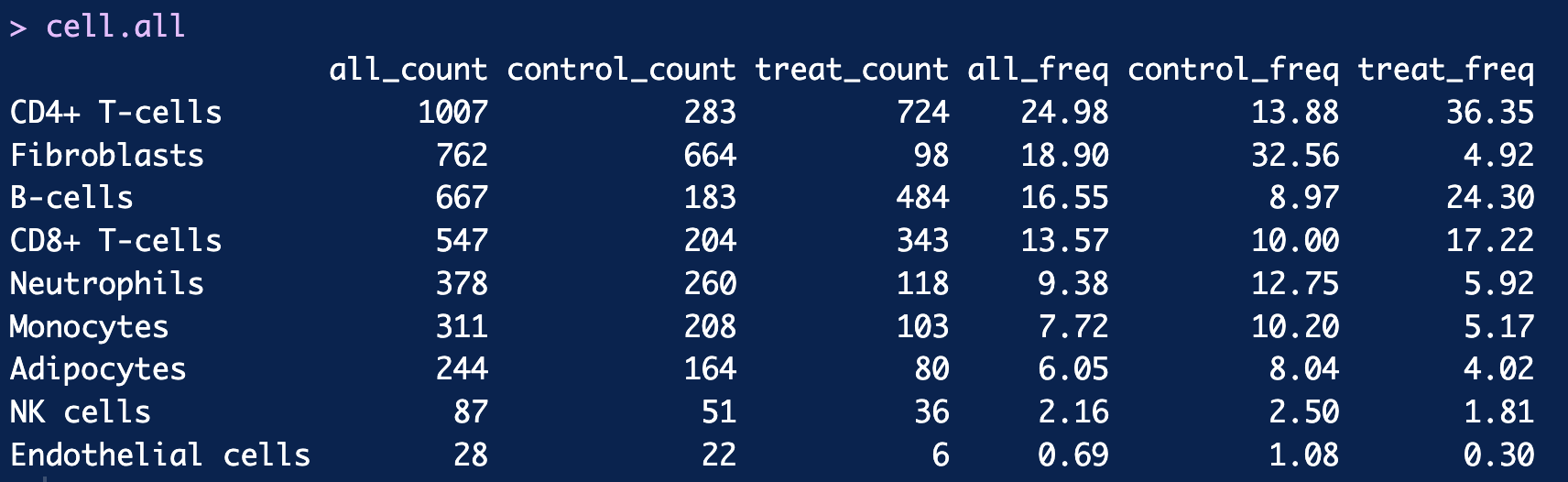
# 8. Calculate the cell numbers in each cluster and their ratio in total cells
cell_counts <- table(Idents(scRNA))
cell.all <- cbind(cell_counts = cell_counts,
cell_Freq = round(prop.table(cell_counts)*100,2))
cell.num.group <- table(Idents(scRNA), scRNA$group)
cell.freq.group <- round(prop.table(cell.num.group, margin = 2) *100,2)
cell.all = cbind(cell.all,cell.num.group,cell.freq.group)
cell.all = cell.all[,c(1,3,4,2,5,6)]
colnames(cell.all) = paste(rep(c("all","control","treat"),times = 2),
rep(c("count","freq"),each = 3),sep = "_")
cell.all
# 9. Defferencial expression analysis
## 9.1 Look for genes that differ between groups within a cell type
sub.obj = subset(scRNA,idents = "NK cells")
dfmarkers <- FindMarkers(scRNA, ident.1 = "treat", group.by = "group",min.pct = 0.25, logfc.threshold = 0.25,verbose = F)
head(dfmarkers)
## 9.2 Look for conserved marker genes
if(!require("multtest"))BiocManager::install('multtest')
if(!require("metap"))install.packages('metap')
sub.markers <- FindConservedMarkers(scRNA, ident.1 = "NK cells", grouping.var = "group", min.pct = 0.25, logfc.threshold = 0.25,verbose = F)
head(sub.markers)
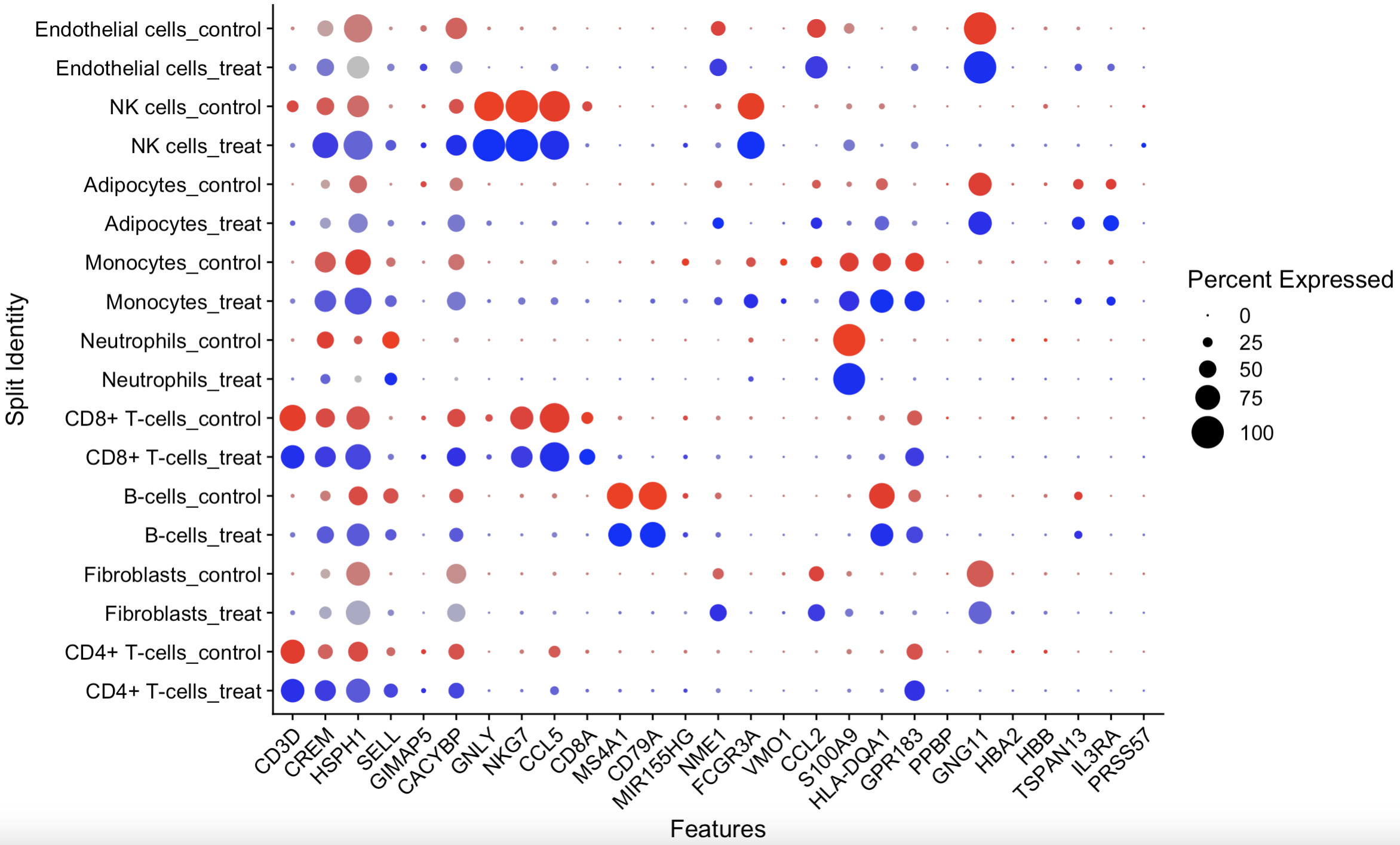
## 9.3 Comparison between groups by Bubble diagram
markers.to.plot = c("CD3D", "CREM", "HSPH1", "SELL", "GIMAP5", "CACYBP", "GNLY", "NKG7", "CCL5",
"CD8A", "MS4A1", "CD79A", "MIR155HG", "NME1", "FCGR3A", "VMO1", "CCL2", "S100A9", "HLA-DQA1",
"GPR183", "PPBP", "GNG11", "HBA2", "HBB", "TSPAN13", "IL3RA", "PRSS57") # a group of interested genes
#scRNA <- subset(scRNA, seurat_annotations %in% na.omit(scRNA$seurat_annotations))
DotPlot(scRNA, features = markers.to.plot, cols = c("blue", "red"),
dot.scale = 8, split.by = "group") +
RotatedAxis()
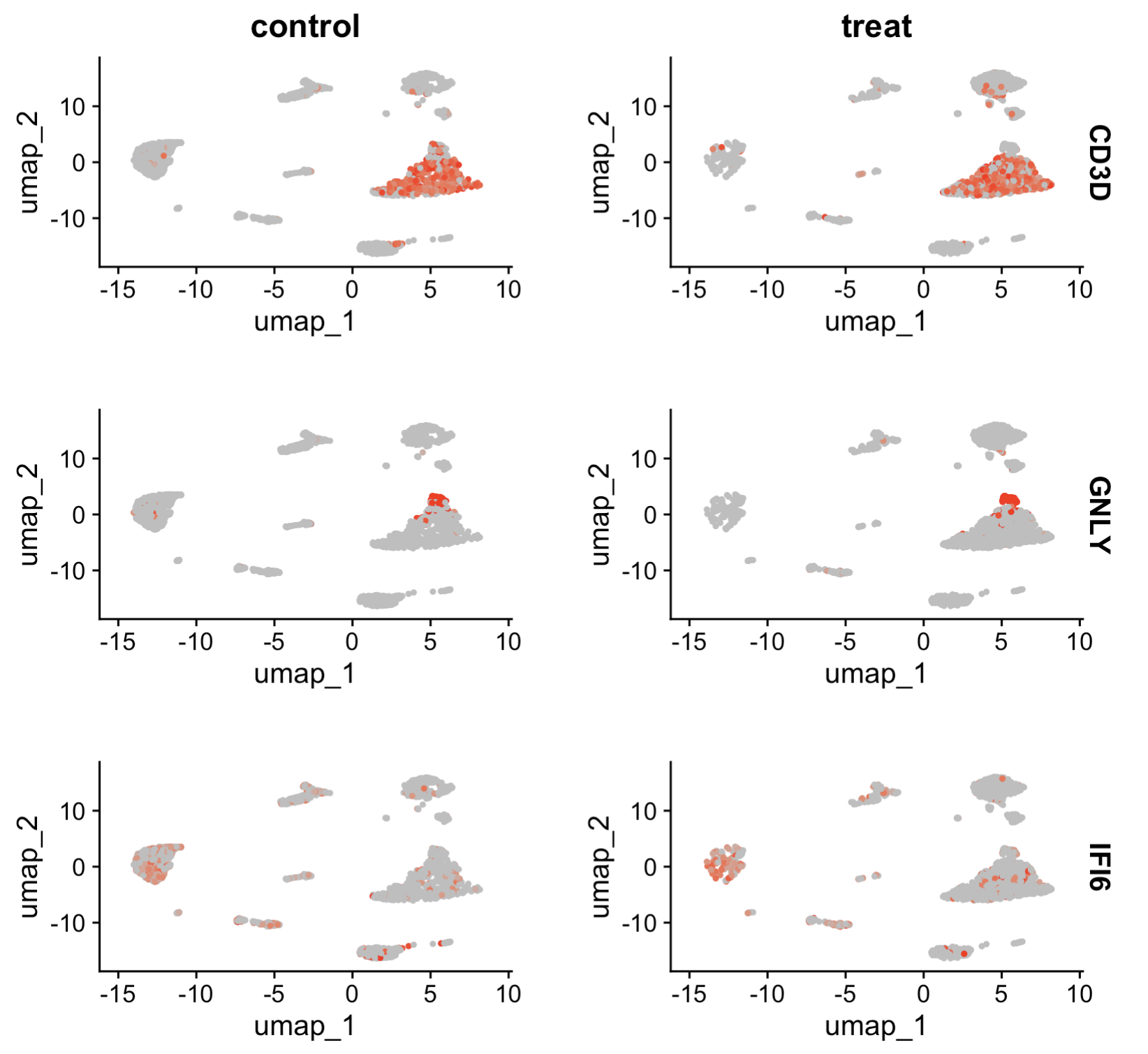
## 9.4 Feature plot
FeaturePlot(scRNA, features = c("CD3D", "GNLY", "IFI6"), split.by = "group", max.cutoff = 3, cols = c("grey",
"red"), reduction = "umap")
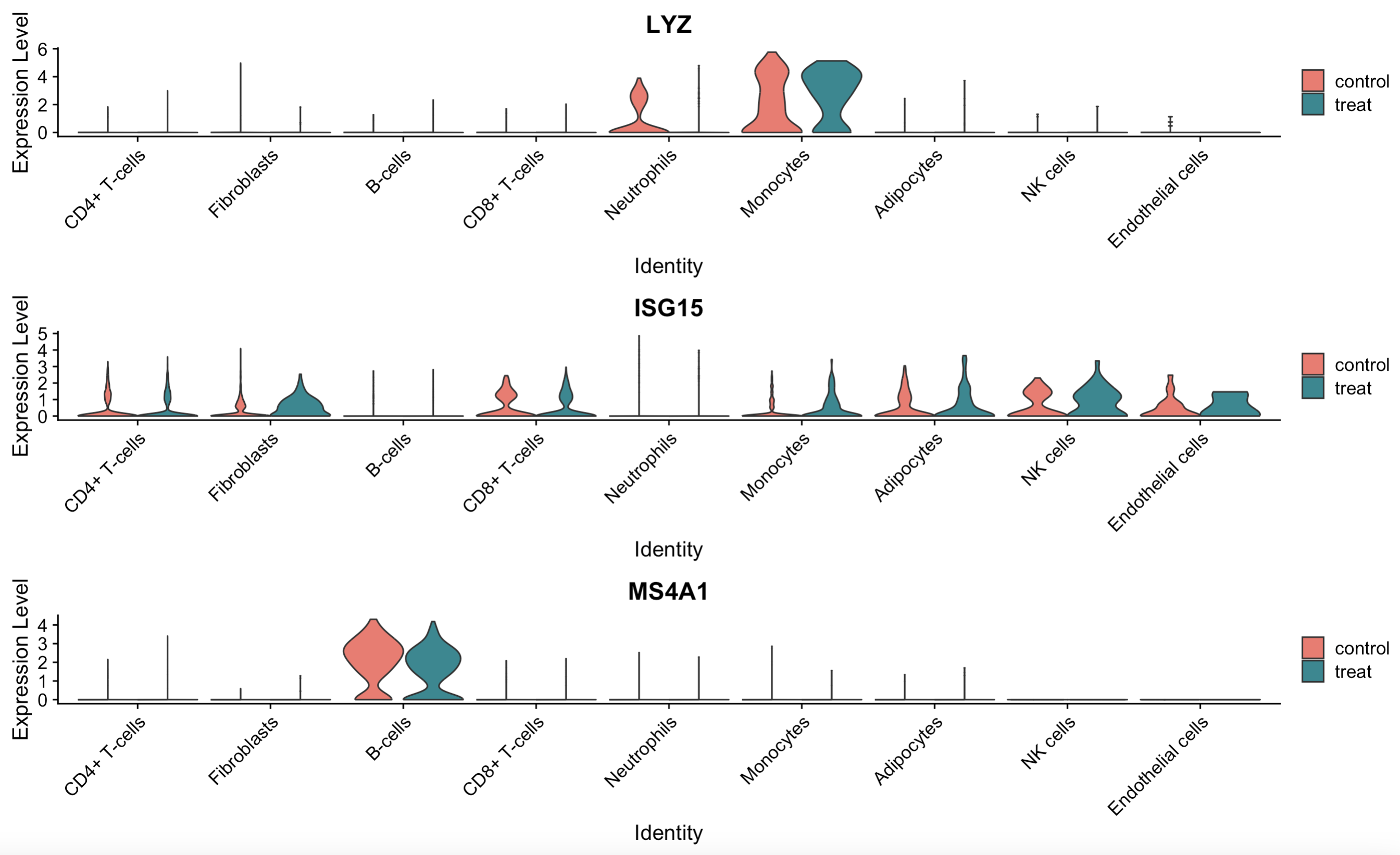
## 9.5 Violin polt
plots <- VlnPlot(scRNA, features = c("LYZ", "ISG15", "MS4A1"), split.by = "group", group.by = "seurat_annotations",
pt.size = 0, combine = FALSE)
library(patchwork)
wrap_plots(plots = plots, ncol = 1)
# 10. Differential analysis of pseudo-bulk transcriptome
## 10.1 Integrate into multiple samples
bulk <- AggregateExpression(scRNA, return.seurat = T, slot = "counts", assays = "RNA", group.by = c("seurat_annotations","orig.ident", "group"))
colnames(bulk)
# AggregateExpression is for integrating scRNA data into bulk RNA-seq data
# group.by = c("seurat_annotations","orig.ident", "group"), one sample is consist of same cell type, same sample, and same grouyp.
# We can also extrac a specific cell type to compare for the DEG between treat and control groups.
sub <- subset(bulk, seurat_annotations == "CD8+ T-cells")
colnames(sub)
Idents(sub) <- "group"
BiocManager::install("DESeq2")
de_markers <- FindMarkers(sub, ident.1 = "treat", ident.2 = "control", slot = "counts", test.use = "DESeq2",
verbose = F)
de_markers$gene <- rownames(de_markers)
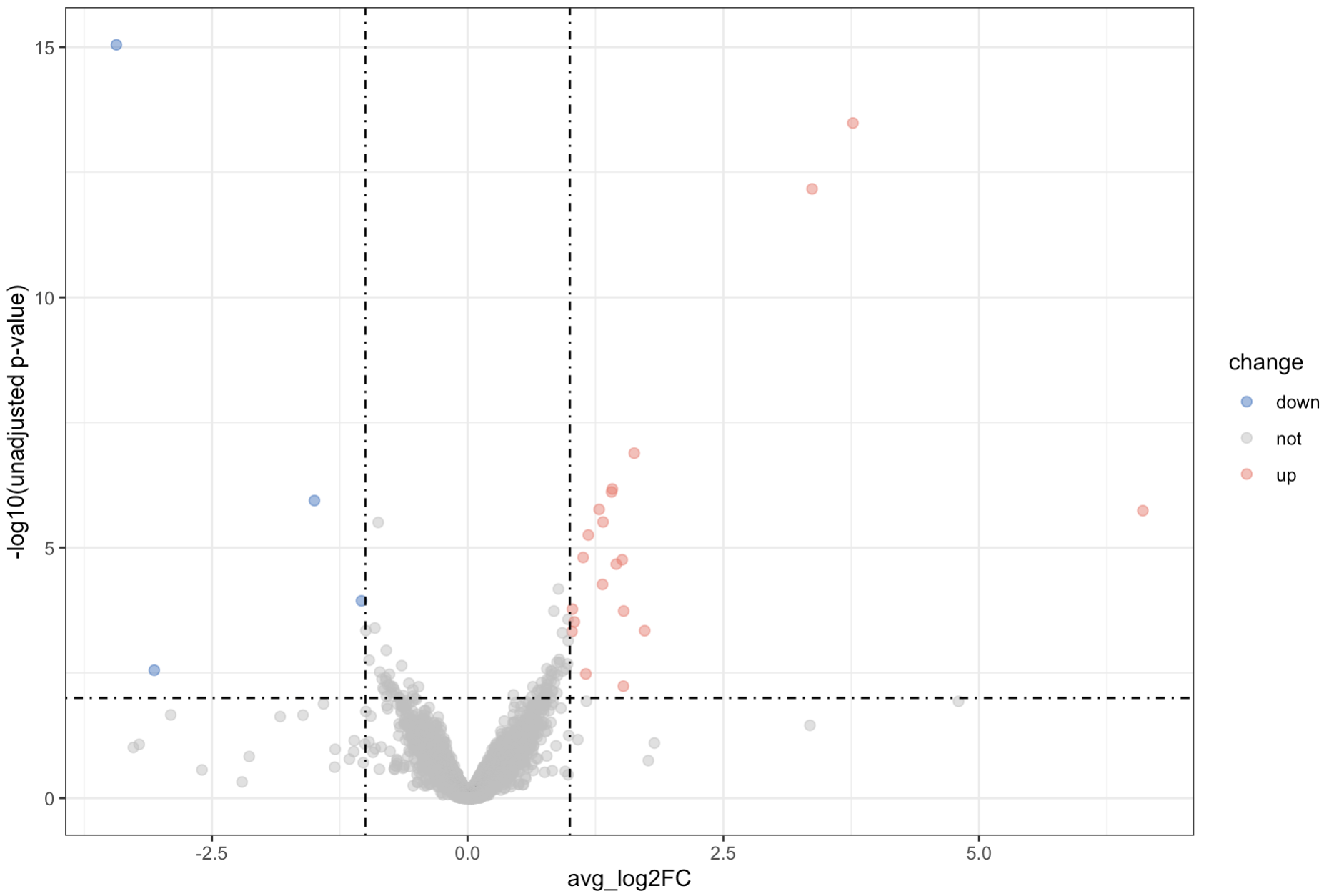
## 10.2 Volcano plot to show DEG
k1 = de_markers$avg_log2FC< -1 & de_markers$p_val <0.01
k2 = de_markers$avg_log2FC> 1 & de_markers$p_val <0.01
de_markers$change <- ifelse(k1,"down",ifelse(k2,"up","not"))
# this is judging for up or down. Satisfy k1 is down, else check for k2, if satisfy k2, is up, if not, is not.
library(ggplot2)
ggplot(de_markers, aes(avg_log2FC, -log10(p_val),color = change)) +
geom_point(size = 2, alpha = 0.5) +
geom_vline(xintercept = c(1,-1),linetype = 4)+
geom_hline(yintercept = -log10(0.01),linetype = 4)+
scale_color_manual(values = c("#2874C5", "grey", "#f87669"))+
theme_bw() +
ylab("-log10(unadjusted p-value)") 4. Quality control by Violin plot

5. Integration of multiple samples by "harmony"
5.1 Elbow plot
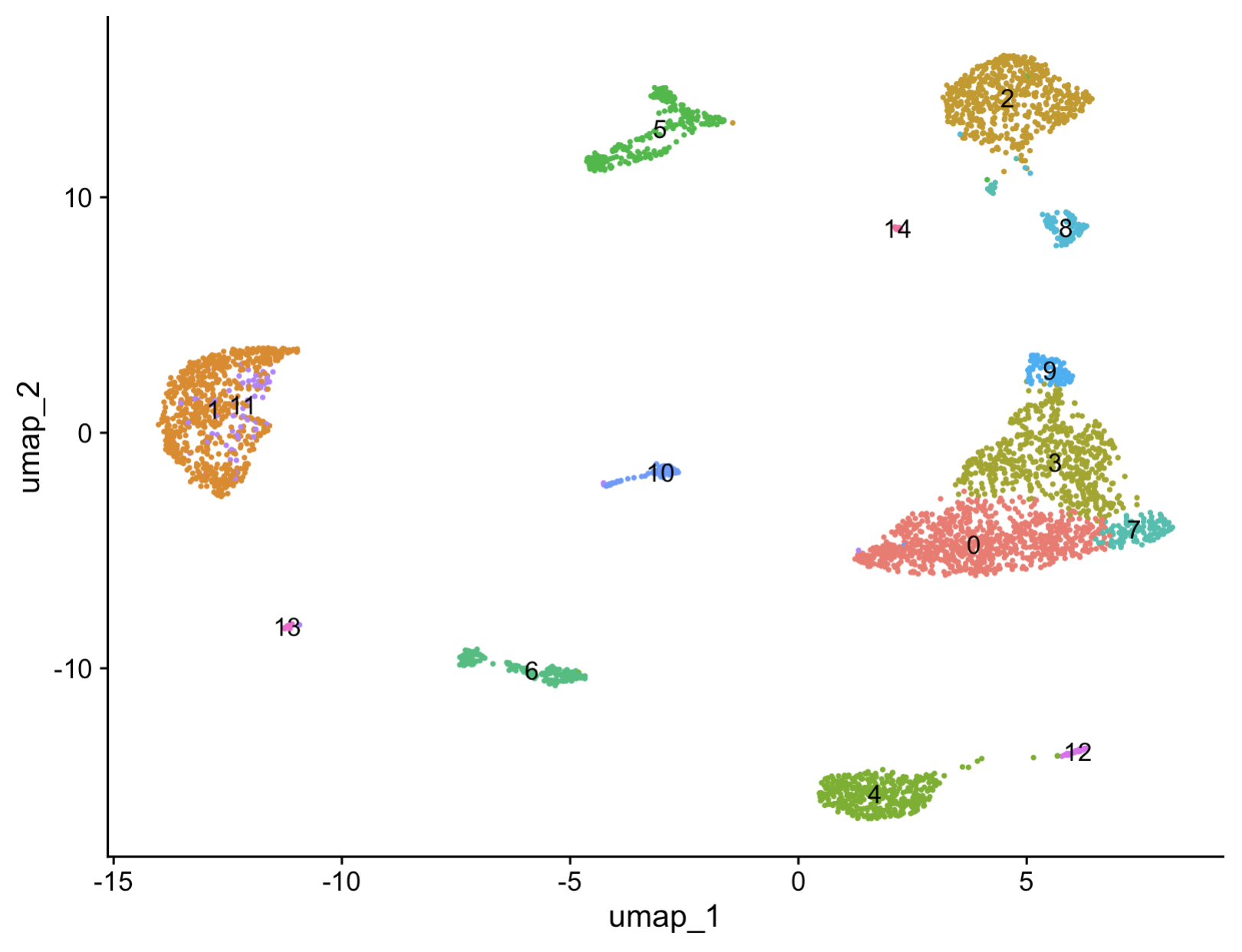
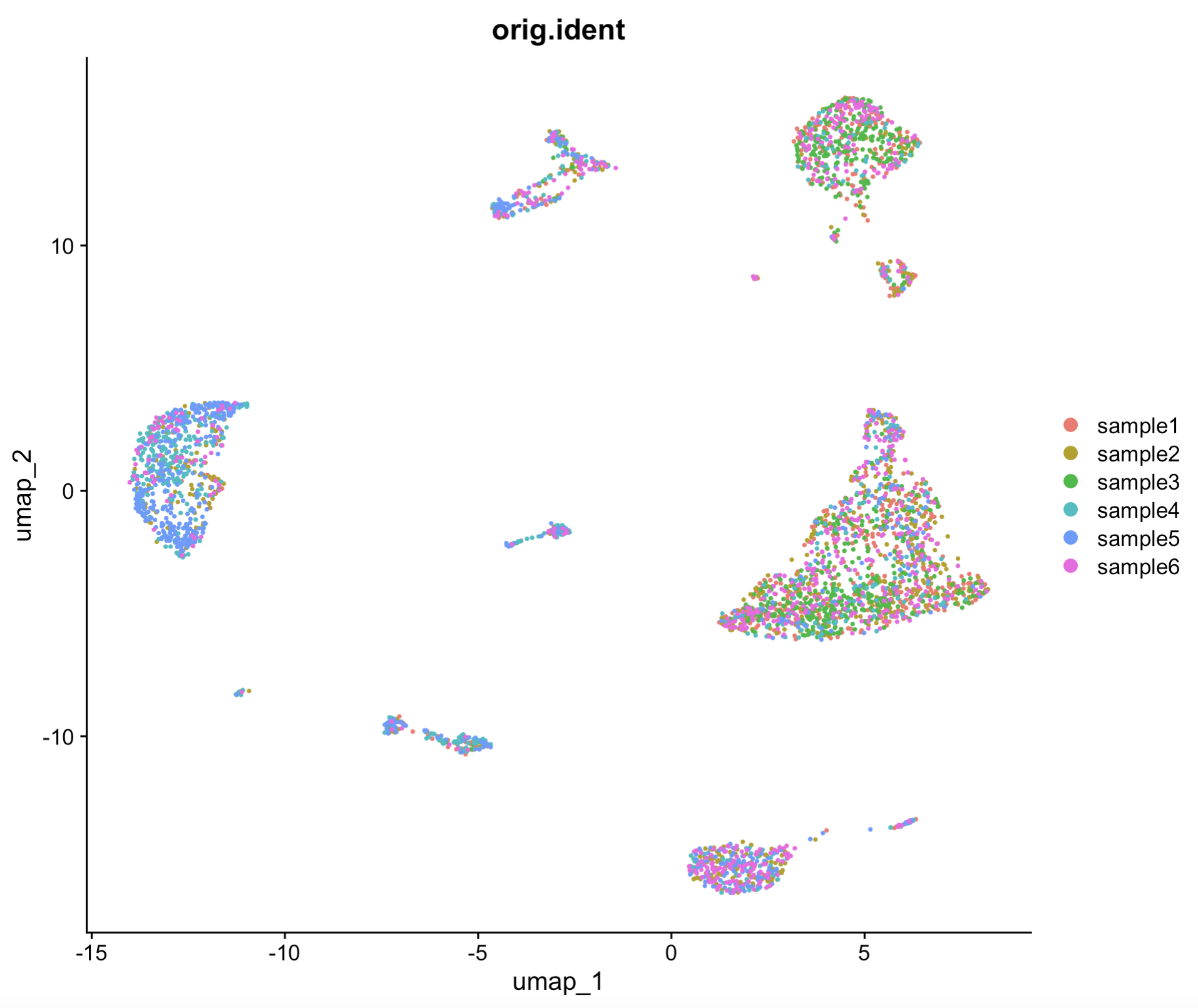
6. Cell type annotation
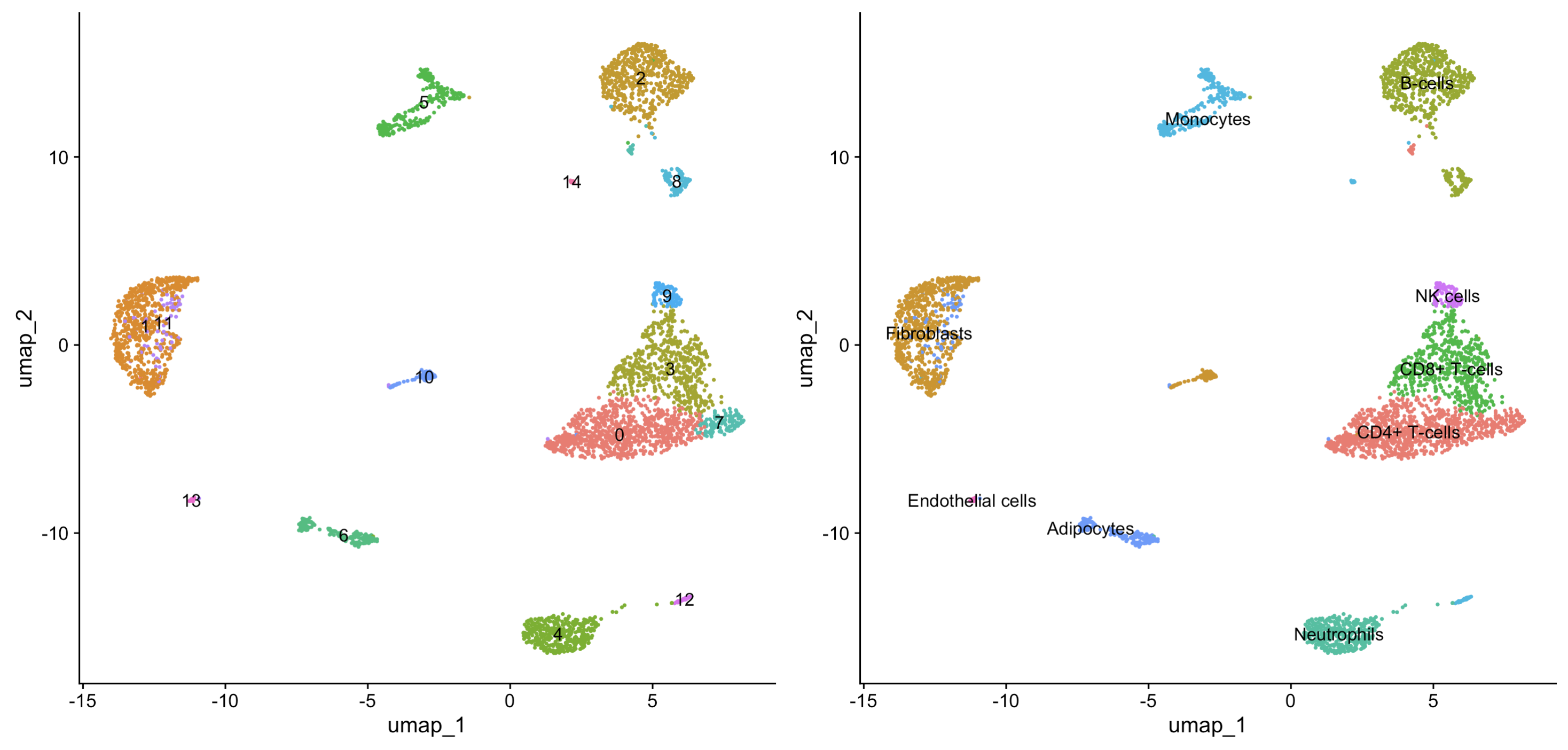
7. Visualisation of grouping and comparisons of cell composition between groups
7.1 Check the grouping information
7.2 Add the group information as a new column, note them on DimPlot
8. Calculate the cell numbers in each cluster and their ratio in total cells
9. Defferencial expression analysis
9.1 Look for genes that differ between groups within a cell type
9.3 Bubble diagram for comparison between groups
9.4 Feature plot
9.5 Violin plot
10.2 Volcano plot for DEGs
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号