如何获得KEGG中某一条通路的全部基因?
原创

如何获得KEGG中某一条通路的全部基因?
比如说我们想看PI3K-Akt signaling pathway这条通路中包含哪些基因?首先需要访问KEGG的官网,获取其hsa编号,如PI3K-Akt signaling pathway对应hsa04151.
访问如下网址(更改对应物种、编号即可),这将返回该通路涉及的所有 人类基因(hsaXXXXX)。KEGG hsa:XXXXX 这个编号通常是 NCBI Entrez Gene ID,即 NCBI 基因数据库 中的唯一基因编号。将网页内容复制到本地。再将Entrez Gene ID转化成基因名即可。
https://rest.kegg.jp/link/hsa/path:hsa04151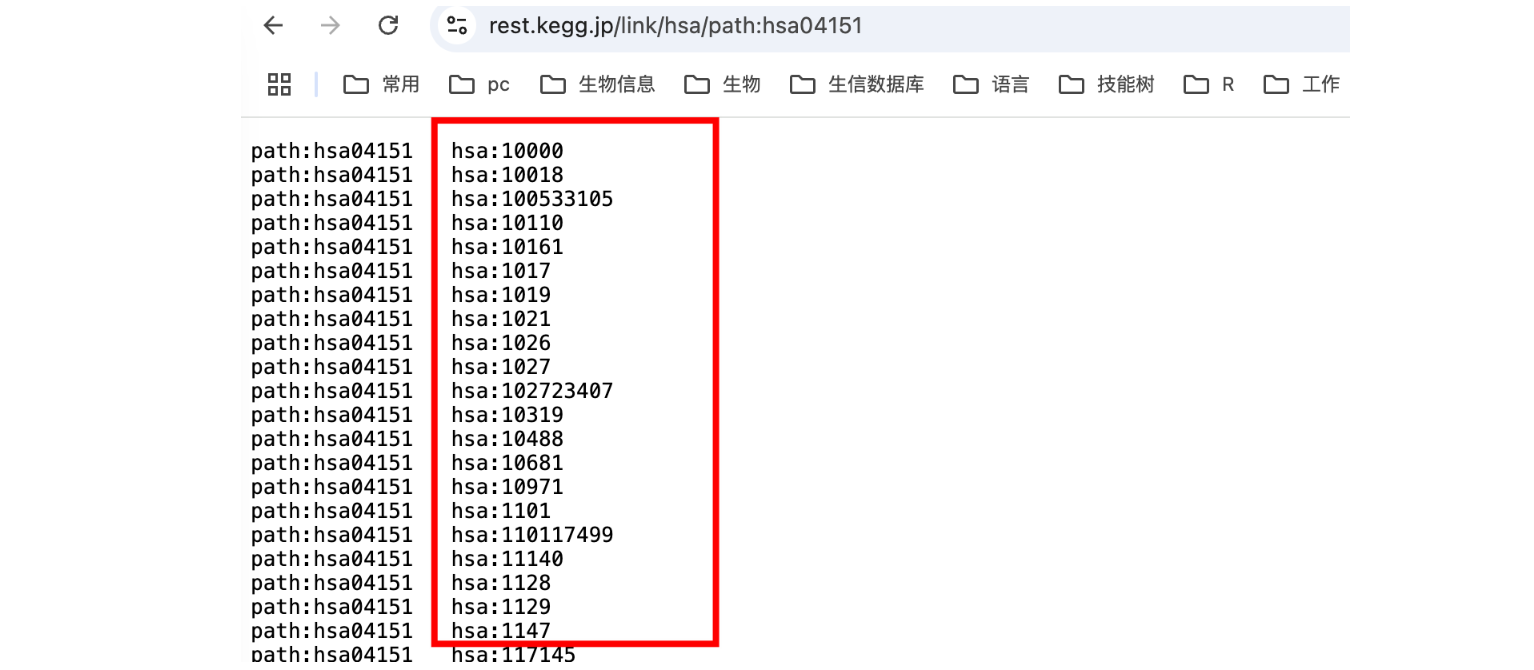
将代码包装成一个函数,方便调用
library(data.table)
library(AnnotationDbi)
library(org.Hs.eg.db)
convert_entrez_to_symbol <- function(file_path) {
# 读取文件
hsa04151_df <- fread(file_path, data.table = FALSE, header = FALSE)
# 提取 ENTREZID
hsa04151_df$ENTREZID <- sub("hsa:", "", hsa04151_df$V2)
# 获取 ENTREZID 到 Gene Symbol 的映射
e2s <- AnnotationDbi::select(org.Hs.eg.db,
keys = hsa04151_df$ENTREZID,
columns = "SYMBOL",
keytype = "ENTREZID")
# 处理缺失值并去重
ids <- na.omit(e2s)
ids <- ids[!duplicated(ids$SYMBOL), ]
ids <- ids[!duplicated(ids$ENTREZID), ]
# 合并数据框
hsa04151_df1 <- merge(hsa04151_df, ids, by = "ENTREZID", all.x = TRUE)
return(hsa04151_df1)
}
# 使用示例
result_df <- convert_entrez_to_symbol("hsa04151.txt")
head(result_df)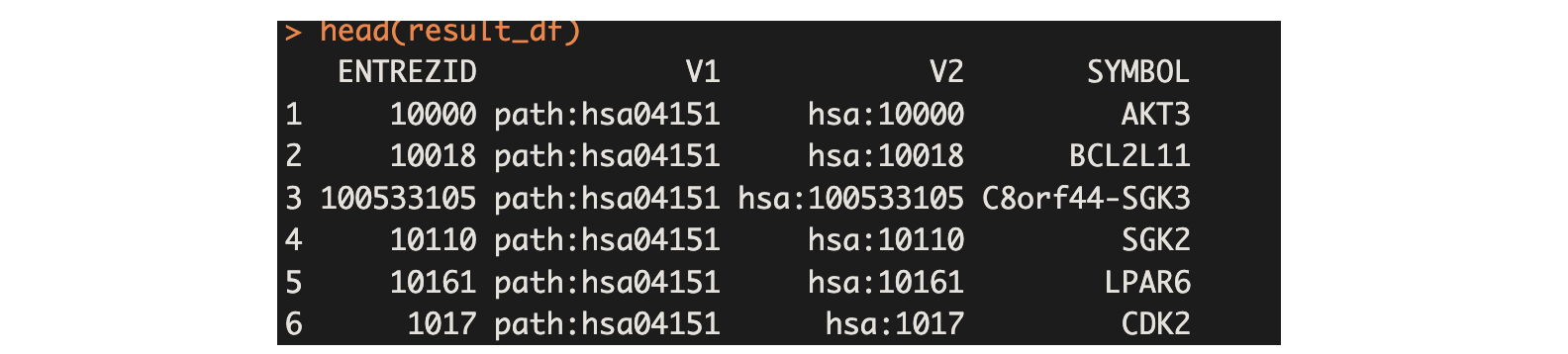
即SYMBOL列中内容,为PI3K-Akt signaling pathway这条通路中所包含的全部基因。
如果存在多个通路,可以执行以下代码
dir("hsa/")
[1] "hsa04010.txt" "hsa04060.txt" "hsa04062.txt" "hsa04151.txt" "hsa04310.txt"
[6] "hsa04330.txt" "hsa04340.txt" "hsa04350.txt" "hsa04380.txt" "hsa04390.txt"
[11] "hsa04510.txt"
library(dplyr)
dir("hsa/")
library(dplyr)
# 获取目录中的所有文件
file_list <- list.files("hsa/", full.names = TRUE)
# 使用 lapply() 依次读取文件并执行函数
result_list <- lapply(file_list, convert_entrez_to_symbol)
# 合并所有返回的结果
final_result <- bind_rows(result_list)
# 查看合并后的结果
head(final_result)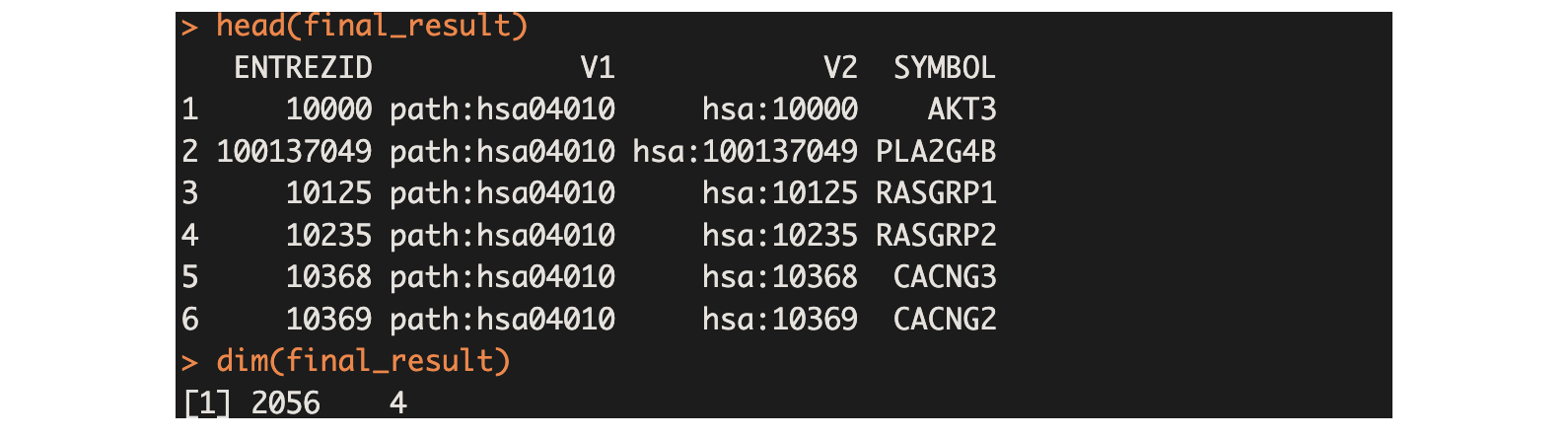
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号