bioRxiv | COATI-LDM:在预训练嵌入空间中雕刻药物分子

bioRxiv | COATI-LDM:在预训练嵌入空间中雕刻药物分子

MindDance
发布于 2026-01-22 12:19:06
发布于 2026-01-22 12:19:06
药物设计不是单目标优化,而是在效力、选择性、生物利用度、安全性等多个维度上的微妙平衡。更棘手的是,ADMET 数据获取成本极高,往往只有数千个样本,远不足以训练复杂的深度学习模型。来自 Terray Therapeutics 的研究团队提出 COATI-LDM,将潜扩散模型应用于由 2.5 亿 分子预训练的嵌入空间,而非直接在分子结构上扩散。通过解耦表示学习和属性优化,COATI-LDM 在 QED 优化基准上达到 95.6% 的成功率,刷新 SOTA 记录。更独特的是引入偏好对齐机制,让生成的分子符合药物化学家的审美直觉。这项发表在 bioRxiv 的工作,为小数据场景下的多参数药物优化提供了新思路。
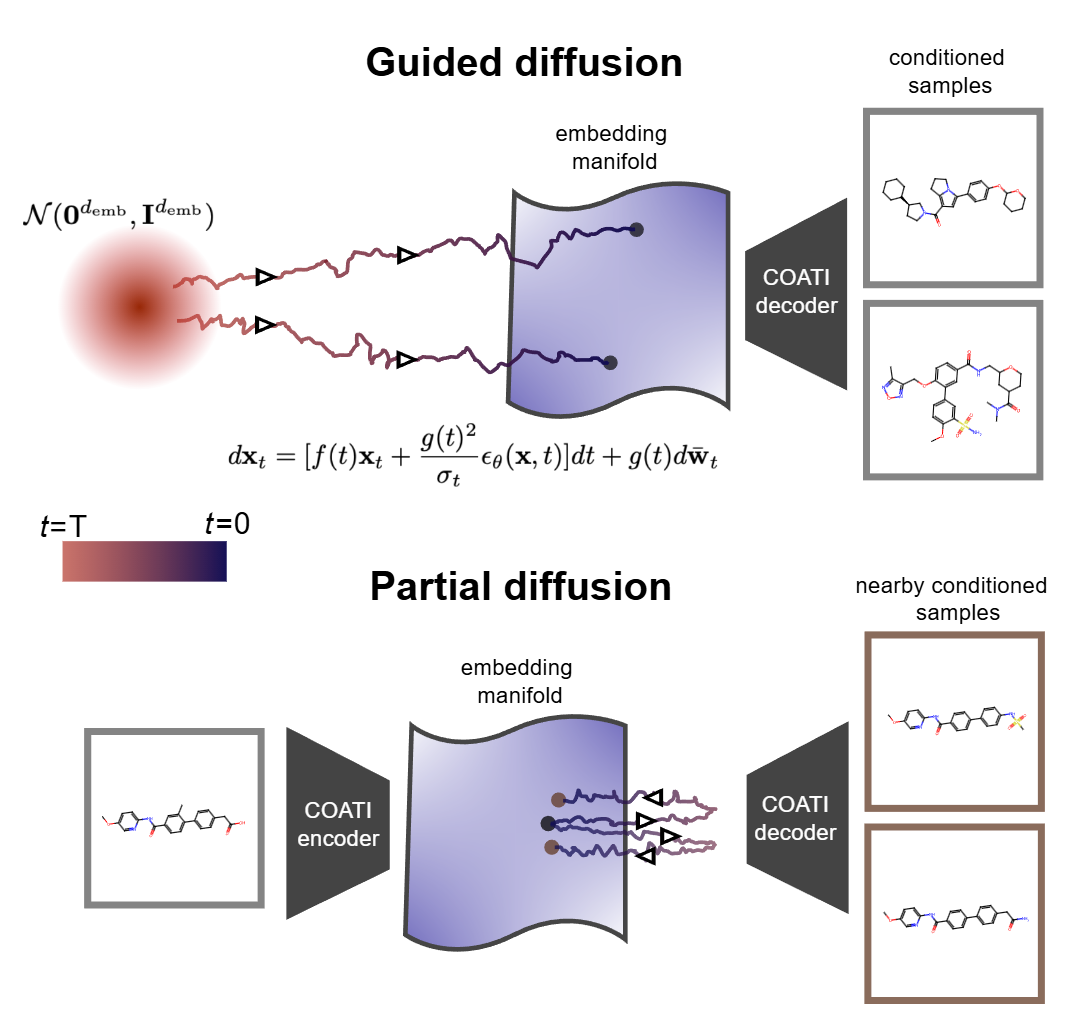
潜扩散与部分扩散的示意图,展示从噪声到分子流形的映射过程
潜扩散与部分扩散的示意图,展示从噪声到分子流形的映射过程
一、多参数优化的困境
ADMET 的数据瓶颈
在药物发现流程中,最昂贵的环节往往不是合成,而是生物测试。一个化合物的吸收、分布、代谢、排泄和毒性(ADMET)评估,可能需要数十种不同的实验,从体外细胞测试到动物模型,每个数据点的成本可能高达数千美元。
这导致了一个严峻的现实:ADMET 数据集极其稀缺。公开数据库如 ChEMBL 虽然包含数百万个化合物的生物活性数据,但针对特定 ADMET 属性的高质量标注往往只有几千到几万条。对于工业界的专有数据,情况更加极端——一个特定靶点的内部测试数据可能少于千例。
在这种小数据场景下,传统的端到端深度学习方法陷入困境。直接在分子图或 3D 坐标上训练扩散模型,需要大量样本来学习化学空间的复杂分布。当训练集只有几千个分子时,模型容易过拟合到训练集的特定区域,泛化能力严重受限。
多目标的平衡艺术
更复杂的是,药物设计需要同时优化多个相互制约的目标。高效力往往伴随高脂溶性,但高脂溶性又可能导致代谢不稳定和毒性风险。增加极性基团能改善溶解度,但可能损害膜透性。这种多参数优化(MPO)问题没有唯一解,而是需要在高维性质空间中找到帕累托前沿。
现有的生成模型在处理多目标约束时表现不佳。遗传算法虽然直观,但需要手工设计化学算子,且在高维空间中搜索效率低下。强化学习可以通过奖励函数引导优化,但每次策略更新都需要大量预言机调用,在 ADMET 这种昂贵的评估场景中不切实际。
二、解耦的智慧
表示学习与属性优化的分离
COATI-LDM 的核心洞察是:不要试图在小数据集上同时学习分子表示和属性优化。相反,将这两个任务解耦——先用海量无标注数据学习通用的分子表示,再用少量标注数据在嵌入空间中进行条件生成。
这个思想虽然直观,但实现起来充满挑战。关键问题是:什么样的嵌入空间适合扩散建模?
答案是 COATI(对比式 2D/3D 编码器)。这是一个基于 Transformer 的对比学习模型,在 2.5 亿 个分子的数据集上预训练。它将每个分子编码为一个 512 维 的连续向量,这个向量不仅捕捉了 2D 拓扑结构(通过 SMILES 或分子图),还整合了 3D 构象信息(通过 Allegro 3D 编码器)。
COATI 的训练目标是对比学习:同一分子的不同构象应该在嵌入空间中彼此接近,不同分子应该远离。这种训练策略赋予嵌入空间良好的几何性质——相似的分子在空间中聚集,插值路径对应平滑的化学变换。
手性感知的关键
COATI 的一个关键改进是采用 Allegro 3D 编码器。与标准的 E(3) 等变网络不同,Allegro 对镜像变换敏感,能够区分对映异构体。这对药物设计至关重要——左旋和右旋分子虽然结构相似,但生物活性可能截然不同。
在嵌入空间的可视化中可以看到,手性分子的两个对映体被映射到不同的区域,证明模型确实学到了立体化学信息。这为后续的属性优化提供了更精细的控制基础。
DirectCLR 避免维度塌陷
对比学习的一个常见陷阱是维度塌陷——模型为了最大化相似样本的一致性,可能将所有表示压缩到低维子空间,损失了表达能力。COATI 采用 DirectCLR 策略,通过正则化协方差矩阵来鼓励使用所有嵌入维度,避免了这个问题。
实验证明,相比标准的对比学习,DirectCLR 训练的嵌入在聚类评估中展现出更好的分离度,意味着化学相似性被更准确地编码。
三、潜空间扩散的精妙设计
在嵌入流形上的扩散
COATI-LDM 在预训练的 512 维嵌入空间中运行扩散过程。前向过程逐步向分子嵌入添加高斯噪声,反向过程则学习去噪,同时以目标属性为条件引导生成方向。
架构采用类似 U-Net 的多层感知机(MLP),但针对分子嵌入的特点进行了调整。时间步嵌入和条件信息通过自适应群归一化(AdaGN)注入到每一层,确保去噪过程持续感知当前的生成目标。
相比直接在分子图上扩散,这个设计带来显著优势。首先,嵌入空间是连续的,扩散过程更加平滑。其次,嵌入维度(512)远低于分子图的表示维度(节点数×特征维度),计算效率更高。最重要的是,预训练嵌入已经包含了丰富的化学知识,模型只需学习如何在这个良好组织的空间中导航。
三种条件引导策略
COATI-LDM 实现了三种条件生成方式:
联合条件是最简单的方法,将目标属性值直接作为额外输入拼接到去噪网络。这种方式计算开销最小,但属性控制精度相对较弱。
分类器引导训练一个独立的回归器预测嵌入对应分子的属性值,然后在扩散过程中沿着属性梯度调整采样方向。这种方式能显著改善属性遵从度——在 hCAII 效力预测中,Pearson 相关系数从联合条件的 0.82 提升到 CG 的 0.94。但代价是样本可能偏离训练数据分布,Frechet 距离(FD)从 0.68 增加到 1.08。
无分类器引导是最优雅的方案。训练时随机丢弃部分条件信息,让模型学会有条件和无条件生成的差异。推理时通过调节条件和无条件预测的加权,实现属性控制与分布匹配的平衡。实验显示,CFG 在改变权重时表现出更好的稳健性,能更好地停留在分子流形上。
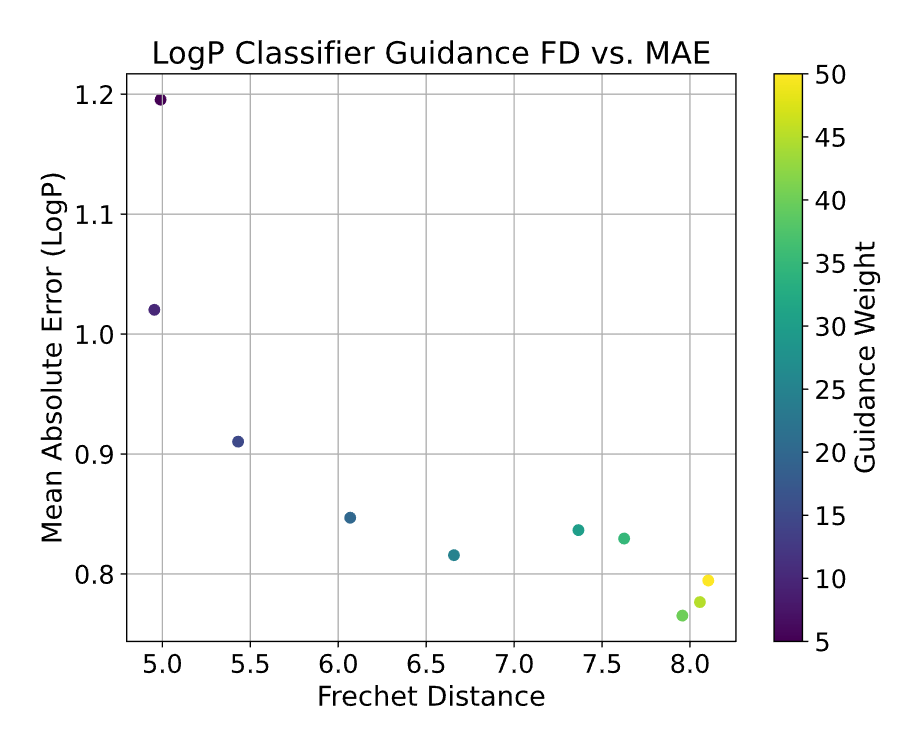
分类器引导权重与 MAE、FD 之间的权衡曲线图
分类器引导权重与 MAE、FD 之间的权衡曲线图
多属性联合控制
在实际药物设计中,往往需要同时优化多个属性。COATI-LDM 支持多属性联合条件作用,实验显示这并不会显著降低对单个属性的控制精度。
例如,同时条件于 hCAII 效力(pIC50)和偏好得分,生成的分子在两个维度上的 MAE 分别为 0.42 和 0.31,与单属性条件相当。这证明嵌入空间的维度足够高,能容纳多个独立的优化方向。
四、局部优化的突破
部分扩散的原理
药物优化的一个常见场景是:已有一个具有合理活性的先导化合物,希望在保留其核心骨架的同时,改善某些不理想的性质(如溶解度、代谢稳定性)。这就是局部优化问题。
COATI-LDM 提出了部分扩散方法来解决这个问题。给定一个种子分子,首先将其编码为嵌入向量 z₀,然后只进行部分加噪到时间步 t(而不是完全扩散到纯噪声),再从 t 时刻开始条件去噪。
这个策略的巧妙之处在于:加噪步数 t 控制了探索范围。t 很小时,只有轻微扰动,生成的分子与种子高度相似;t 较大时,允许更激进的结构变化。通过调节 t,可以在相似性和优化幅度之间精确权衡。
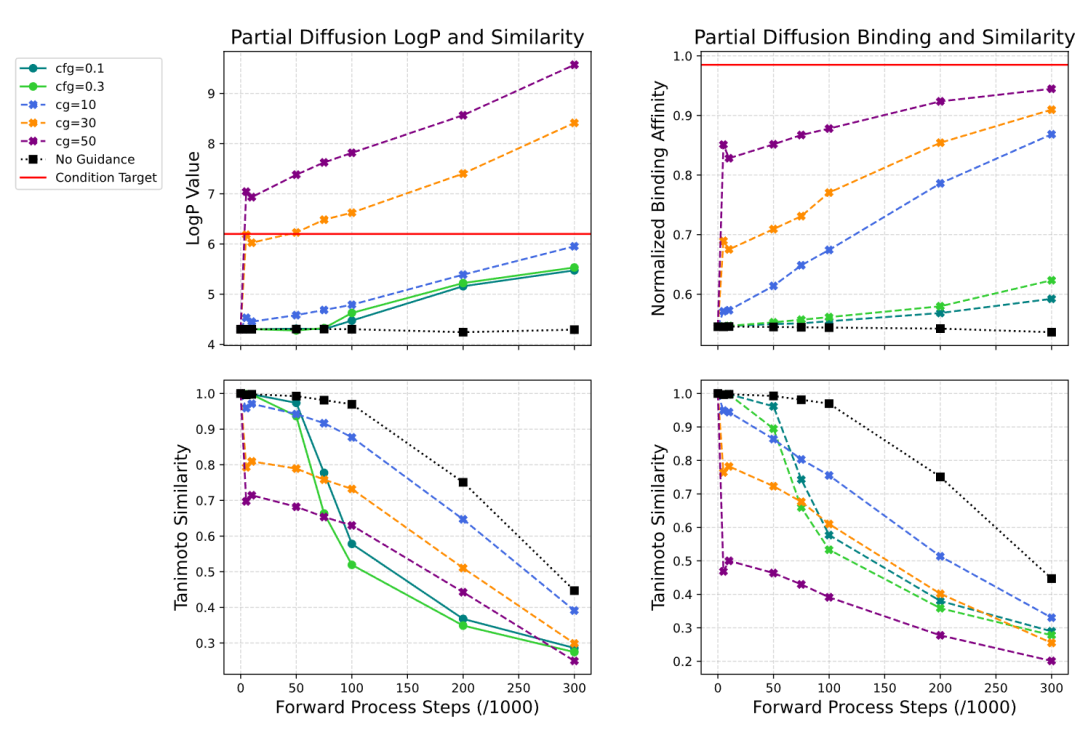
部分扩散中步数与相似度、属性优化值之间的关系曲线
部分扩散中步数与相似度、属性优化值之间的关系曲线
QED 基准的刷新
在经典的 QED(药物相似性)优化基准测试中,任务是将 QED 值从 0.6-0.8 的起始分子优化到 0.9 以上,同时保持 Tanimoto 相似度不低于 0.4。这个基准被广泛用于评估局部优化算法。
COATI-LDM 在这个任务上达到了 95.6% 的成功率,超越了此前的 SOTA 记录(MolMIM 的 94.6%)。更值得注意的是平均相似度保持在 0.48,说明模型不是通过激进的结构重排来提升 QED,而是进行了精细的局部修饰。
案例分析显示,模型倾向于添加羟基、氨基等提升氢键能力的基团,或者调整环系大小来优化分子形状。这些修改策略与药物化学家的直觉高度一致。
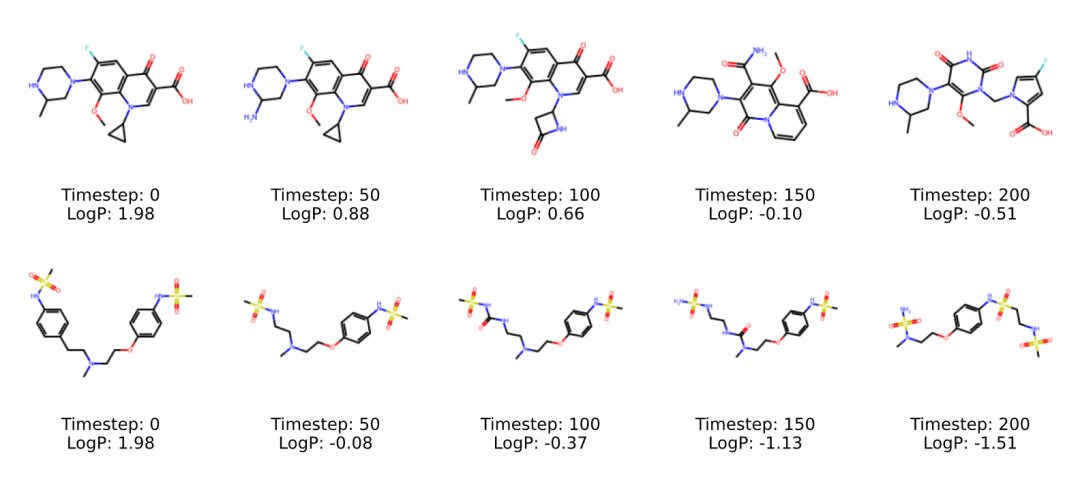
以 Gatifloxacin 等药物为起点的局部优化生成实例
以 Gatifloxacin 等药物为起点的局部优化生成实例
五、偏好对齐的探索
捕捉专家直觉
除了可量化的属性如 logP、TPSA,药物化学家在评估分子时还依赖大量隐性知识——某些官能团组合虽然通过了所有计算筛选,但经验表明它们在合成或后续优化中会遇到问题。
COATI-LDM 引入了偏好学习机制来捕捉这种专家直觉。研究团队邀请药物化学家对 5000 对分子进行成对比较,标注哪个更符合先导化合物的标准。这些数据用于训练一个 RankNet 模型,学习将分子嵌入映射到偏好得分。
训练好的 RankNet 作为额外的条件信号整合到扩散过程中。生成时,可以要求模型产出高偏好分子,引导结果向专家认可的化学空间倾斜。
验证与局限
为了验证偏好模型的有效性,研究团队进行了盲测。生成两批分子——一批以高偏好为目标,一批以低偏好为目标,然后让新的药物化学家(未参与训练数据标注)进行评估。
结果显示,76.8% 的情况下,专家更倾向于模型标记为高偏好的分子。这个比例虽然不是压倒性的,但考虑到化学审美的主观性,已经证明模型确实学到了有意义的模式。
但偏好模型也暴露出局限。论文展示了一个失败案例:模型给一个包含高度紧张环系统的分子打了高分,但这个分子在化学上极不稳定。这揭示了当前偏好模型对底层物理约束的理解不足——它学到了表面的结构模式,但缺乏对化学稳定性的深层认知。
RankNet 模型推断的偏好得分分布及案例分析
RankNet 模型推断的偏好得分分布及案例分析
六、实战检验
hCAII 靶点的覆盖度
人类碳酸酐酶 II(hCAII)是一个经典的药物靶点。研究团队使用内部的 hCAII 结合亲和力数据训练 COATI-LDM,然后评估生成分子对已知活性化合物的覆盖度。
结果令人印象深刻。在 Tanimoto 相似度阈值 0.7 下,CFG 模型对内部持有数据的覆盖率达到 *28.6%*,而遗传算法几乎为零。这意味着,通过条件生成,模型成功重新发现了超过四分之一的已知活性分子或其结构类似物。
对于公开数据集中的 hCAII 活性分子,CG 方法表现更好,覆盖率达到 1.1%(遗传算法仅 0.6%)。虽然绝对数值较低,但考虑到化学空间的广阔性,这已是显著成就。
更有意思的是,研究团队发现 CFG 和 CG 在覆盖的分子类型上存在互补性。CFG 更擅长捕捉训练集主体分布的特征,而 CG 能探索到分布边缘的孤立活性簇。这提示在实际应用中,可以结合多种引导策略来最大化化学空间的探索。
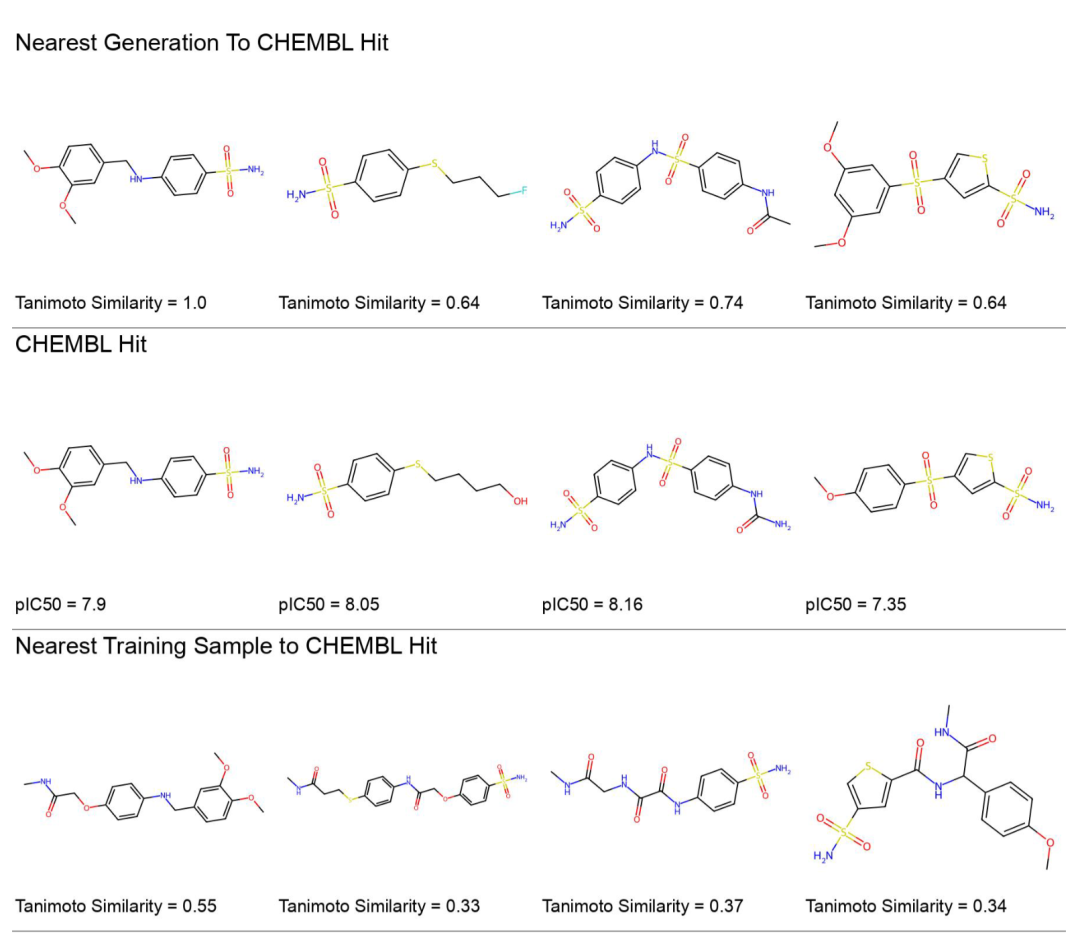
生成的 hCAII 分子及其与公共库中已知分子的相似度对比
生成的 hCAII 分子及其与公共库中已知分子的相似度对比
粒子引导的多样性提升
标准的扩散采样每次独立生成,但在多参数优化场景中,我们希望一次生成就覆盖帕累托前沿的多个区域。COATI-LDM 实现了粒子引导机制——在去噪过程中,让多个粒子(样本)相互排斥,鼓励它们占据嵌入空间的不同区域。
t-SNE 可视化显示,引入粒子引导后,生成样本的分布从单一簇扩展到多个离散簇,显著提升了结构多样性。这对于需要快速探索多种设计策略的早期药物发现尤其有价值。
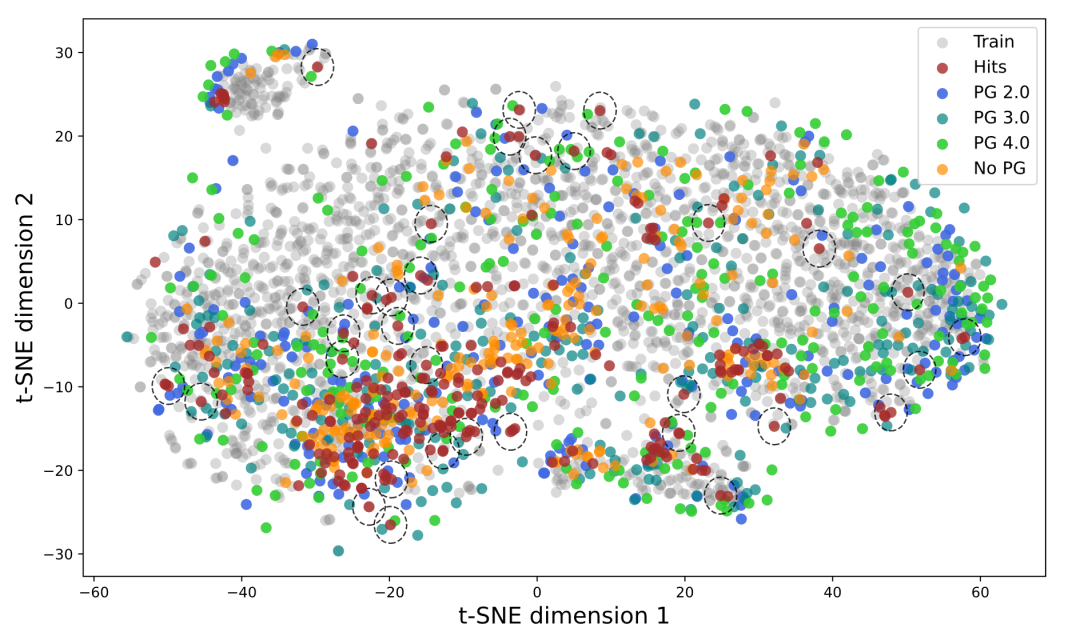
展示粒子引导如何增加生成样本的多样性并覆盖孤立的活性簇
展示粒子引导如何增加生成样本的多样性并覆盖孤立的活性簇
与自回归 Transformer 的对比
为了公平比较,研究团队训练了一个自回归 Transformer 作为基线,该模型在 263M 分子的数据集上预训练,然后在相同的 hCAII 数据上微调。
在单属性控制任务(如 TPSA、fsp3)中,扩散模型仅用 0.48M 数据训练就达到了与 Transformer 相当的 RMSE(fsp3 的 RMSE,扩散模型约 0.08,Transformer 约 0.06)。在多属性联合控制下,扩散模型表现出更好的稳定性——Transformer 在处理相互冲突的约束时,某些属性的误差会显著增大。
这个对比揭示了扩散模型的一个独特优势:在潜空间中,多个条件可以通过线性组合自然地整合,而自回归模型的序列生成机制使得全局协调多个约束更加困难。
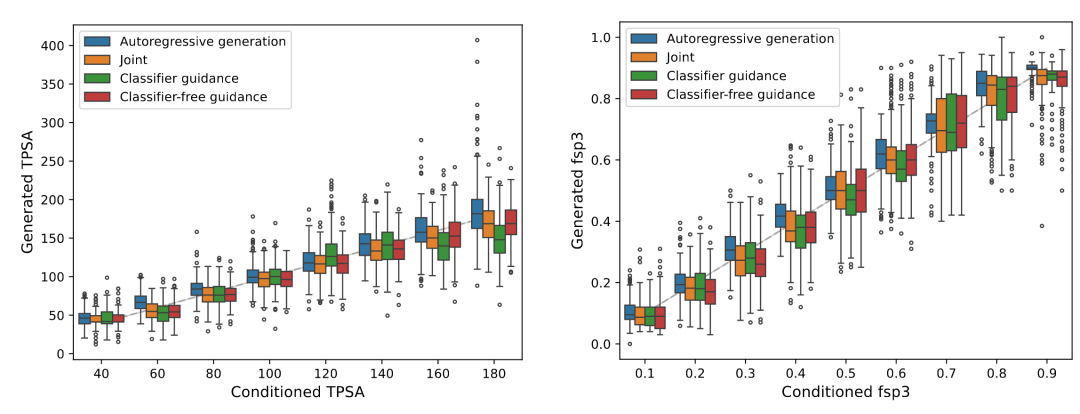
箱线图,对比 Transformer 与扩散模型在 TPSA 和 fsp3 上的控制精度
箱线图,对比 Transformer 与扩散模型在 TPSA 和 fsp3 上的控制精度
七、深层的洞察
预训练嵌入的价值
COATI-LDM 的成功本质上验证了迁移学习的威力。在计算机视觉中,先在 ImageNet 上预训练,再在小数据集上微调,已是标准范式。但在化学领域,这个范式的应用并不充分。
COATI 提供的 512 维嵌入,可以看作分子的通用特征提取器。它在 2.5 亿分子上学到的化学模式——官能团的分布、骨架的拓扑、3D 构象的偏好——为下游的属性优化提供了坚实基础。当标注数据只有几千条时,不需要从头学习这些基础知识,只需学习如何在已有的表示空间中导航。
多模态融合的未来
COATI 同时整合 2D 和 3D 信息,这是一个重要的设计选择。纯 2D 方法(如基于 SMILES 的模型)无法捕捉立体化学;纯 3D 方法(如基于坐标的扩散)计算成本高且难以处理柔性分子的多构象问题。
对比学习提供了一个优雅的融合方案:让同一分子的 2D 拓扑和 3D 构象在嵌入空间中对齐。这种隐式融合比显式的特征拼接更灵活,模型能自动学习何时依赖 2D 信息(如对于平面芳香环),何时依赖 3D 信息(如对于手性中心)。
可控性与可解释性
扩散模型的一个优势是条件控制的透明性。通过调节引导权重,可以直观地观察属性控制强度与分布匹配度的权衡。相比之下,在强化学习框架中,奖励函数的设计往往是试探性的,很难预测最终策略的行为。
偏好学习更进一步提升了可解释性。通过分析 RankNet 的注意力权重,可以识别出哪些结构特征与高偏好相关。虽然这不等同于因果解释,但为药物化学家提供了检查模型决策逻辑的窗口。
八、局限与展望
尽管 COATI-LDM 在多个方面取得突破,仍有改进空间。当前的偏好模型仅在 5000 对样本上训练,对化学稳定性的理解不够深入。未来可以引入主动学习策略,让模型主动询问药物化学家对不确定样本的评价,迭代改进偏好函数。
MLP 架构虽然简单有效,但未必是分子嵌入空间的最优选择。未来可以探索专门为向量数据设计的得分网络,如基于注意力机制的 Transformer 变体,或者利用嵌入空间的几何性质设计等变网络。
从应用角度看,将 COATI-LDM 整合到闭环优化系统是自然的下一步。想象一个工作流:模型生成候选分子 → 实验测试 → 反馈数据用于微调 → 生成下一批候选。这种人机协同的主动学习循环,有望大幅提升药物发现的效率。
更宏大的愿景是多目标帕累托优化。当前的条件生成虽然能处理多个属性,但主要是按给定目标值生成。理想情况下,应该能生成帕累托最优解的集合——即在某一属性上无法改进而不牺牲其他属性的分子。这需要将扩散模型与多目标优化算法(如 NSGA)结合,是一个充满挑战的研究方向。
值得思考的问题
Q1: 为什么在嵌入空间扩散比在分子图上扩散更高效?
嵌入空间是连续的低维流形,而分子图是离散的高维对象。在嵌入空间中,每一步扩散都是平滑的向量加法,计算开销仅取决于嵌入维度(512);在分子图上,每一步需要更新节点和边的离散特征,计算复杂度与图大小成正比。更重要的是,嵌入空间已经通过预训练编码了化学知识,扩散过程自动继承了这些约束,而图扩散需要从头学习化学规则。
Q2: 部分扩散如何在相似性和优化幅度之间权衡?
部分扩散的关键参数是加噪步数 t。t 较小时,只有高频细节被破坏,去噪过程只能进行局部修饰,相似度高但优化空间有限;t 较大时,骨架结构也被扰动,去噪有更大自由度重构分子,能实现更激进的优化但相似度降低。实验显示,在 t=20 附近(总步数 1000),能达到 Tanimoto 相似度 0.4-0.5 且 QED 提升 0.2-0.3 的良好平衡。
Q3: 偏好学习如何避免编码错误的偏见?
这是一个开放问题。当前的偏好模型完全依赖人类标注,如果标注者存在系统性偏见(如过度偏好某类骨架),模型会忠实地学习并放大这些偏见。缓解策略包括:增加标注者多样性、引入客观的稳定性检查(如量子化学计算)、通过主动学习识别模型不确定的区域并重点标注。最终目标是构建一个结合人类直觉和物理约束的混合偏好函数。
Q4: 如何理解扩散模型在多属性控制上优于自回归模型?
自回归模型按顺序生成 token,每个决策只能看到之前的历史,难以进行全局协调。当有多个冲突的属性约束时(如高极性与高膜透性),模型在生成序列的某个位置可能被迫做出妥协,后续难以纠正。扩散模型的去噪过程是并行的——每一步更新整个分子嵌入,多个条件信号可以通过梯度叠加自然融合。这种全局优化机制更适合处理多参数约束。
参考文献:Kaufman B, Williams E C, Pederson R, et al. Latent diffusion for conditional generation of molecules[J]. bioRxiv, 2024: 2024.08. 22.609169.
代码数据:
https://github.com/terraytherapeutics/COATI-LDM
欢迎在评论区分享您对本工作的看法。如果您对 AI 药物发现和多参数优化感兴趣,欢迎关注 MindDance 公众号获取更多前沿内容,也欢迎将文章分享给您的同行。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-01-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号