CutRun and CutTag——Linux篇


最近课题有用到Cut&Tag和Cut&Run,所以这周推文想分享一下Cut&Run上游分析。
Cut &Run 和 Cut & Tag的常规流程多数步骤是一致的,我的全部流程主要参考了三篇教程,在此致谢。
- CUT&Tag Data Processing and Analysis Tutorial (protocols.io)
- 新的ngs流程该如何学习(以CUT&Tag 数据处理为例子)
- #cuttag
Cut &Run 和 Cut & Tag 都是Steven Henikoff实验室开发的方法。
先从linux command入手,来跑一遍CUT RUN的流程,用的数据是实验室测序的数据。
conda下创建一个小环境 cuttag
conda create -n cuttag python=3
source ./activate
conda install -c bioconda -y fastqc bowtie2 samtools bedtools picard deepTools multiqc
conda install -c conda-forge r-base
#SEACR is R package
用的学校的MSI服务器,下面是提交任务脚本。
#!/bin/bash -l
#SBATCH --time=20:00:00
#SBATCH --ntasks=20
#SBATCH --mem=60g
#SBATCH --tmp=20g
#SBATCH --mail-type=ALL
conda activate cuttag
module load python3
module load bowtie2
module load samtools
module load bedtools
projPath="/home/cheny5/han00433/project/Haiping_CutRun_101623"
mkdir -p ${projPath}/fastq
mkdir -p ${projPath}/alignment/sam/bowtie2_summary
mkdir -p ${projPath}/alignment/bam
mkdir -p ${projPath}/alignment/bed
mkdir -p ${projPath}/alignment/bedgraph
mkdir -p $projPath/alignment/sam/fragmentLen
mkdir -p $projPath/peakCalling/SEACR
cores=16
spikeInRef="/home/cheny5/han00433/project/Ecoli/Ecoli"
ref="/home/cheny5/han00433/project/homo_index/GRCh38_noalt_as/GRCh38_noalt_as"
chromSize="/home/cheny5/han00433/project/GRCh38_noalt_as/hg38.chrom.sizes"
seacr="/home/cheny5/han00433/project/SEACR/SEACR_1.3.sh"
histControl= N8_NC
Cutadapt 和整理数据
##== linux command ==##
#我在官方文档中看到,说是不建议cut接头,但是不太适用我们的数据。
#因为我们不cut接头后面比对率就只有百分之十几,cut掉以后有百分之七十多或者八十多。
## bowtie2 GRCh38 $ref
#N7_PC组是postive control: H3K4me3
#N8_NC组是negative control: IgG
for histName in H1_HU H2_HU H3_HU_IgG H4_HU H5_HU H6_HU_IgG H7_HU N1 N2 N1_HU N2_HU N3_HU_IgG N4_HU N5 N5_HU N6 N6_HU_IgG N7_HU N7_PC N8_NC
do
cutadapt -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCA -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT -m 20 -o ${projPath}/fastq/${histName}cut_1.fq.gz -p ${projPath}/fastq/${histName}cut_2.fq.gz ${projPath}/fastq/${histName}_1.fq.gz ${projPath}/fastq/${histName}_2.fq.gz
done
#因为H_HU和N_HU组都各有两个IgG, 想着为了能够得到更多的peak,重复性更好,考虑把两组IgG数据合并。
#H3_HU_IgG H6_HU_IgG
#N3_HU_IgG N6_HU_IgG
cat H3_HU_IgGcut_1.fq.gz H6_HU_IgGcut_1.fq.gz > H_HU_IgGcut_1.fq.gz
cat H3_HU_IgGcut_2.fq.gz H6_HU_IgGcut_2.fq.gz > H_HU_IgGcut_2.fq.gz
cat N3_HU_IgGcut_1.fq.gz N6_HU_IgGcut_1.fq.gz > N_HU_IgGcut_1.fq.gz
cat N3_HU_IgGcut_2.fq.gz N6_HU_IgGcut_2.fq.gz > N_HU_IgGcut_2.fq.gz
bowtie2 GRCh38 and E.coli
在bowtie2 比对的时候需要构建索引
#homo spices 建索引
#下面有建好的索引我就直接用了,因为建索引所需资源比较多,时间长。
https://genome-idx.s3.amazonaws.com/bt/GRCh38_noalt_as.zip
#Escherichia coli str. K-12 substr. MG1655建索引
#https://bacteria.ensembl.org/Escherichia_coli_str_k_12_substr_mg1655_gca_000005845/Info/Index
bowtie2-build Escherichia_coli_str_k_12_substr_mg1655_gca_000005845.ASM584v2.dna.chromosome.Chromosome.fa ecoli
Convert Sam to Bam and Bed
「include the step of spike in」
## bowtie2 GRCh38 $ref
for histName in H1_HU H2_HU H3_HU_IgG H4_HU H5_HU H6_HU_IgG H7_HU N1 N2 N1_HU N2_HU N3_HU_IgG N4_HU N5 N5_HU N6 N6_HU_IgG N7_HU N7_PC N8_NC H_HU_IgG N_HU_IgG
do
## Build the bowtie2 reference genome index if needed:
## bowtie2-build path/to/hg38/fasta/hg38.fa /path/to/bowtie2Index/hg38
echo "$histName Bowtie2 hg38"
echo Job started at `date`
bowtie2 --end-to-end --very-sensitive --no-mixed --no-discordant --phred33 -I 10 -X 700 -p ${cores} -x ${ref} -1 ${projPath}/fastq/${histName}cut_1.fq.gz -2 ${projPath}/fastq/${histName}cut_2.fq.gz -S ${projPath}/alignment/sam/${histName}_bowtie2.sam &> ${projPath}/alignment/sam/bowtie2_summary/${histName}_bowtie2.txt
echo "$histName bowtie2 end"
## Bowtie2 Ecoli
echo "$histName Bowtie2 Ecoli"
echo Job started at `date`
bowtie2 --local --very-sensitive --no-overlap --no-dovetail --no-mixed --no-discordant --phred33 --quiet -I 10 -X 700 -p ${cores} -x ${spikeInRef} -1 ${projPath}/fastq/${histName}cut_1.fq.gz -2 ${projPath}/fastq/${histName}cut_2.fq.gz -S $projPath/alignment/sam/${histName}_bowtie2_spikeIn.sam &> $projPath/alignment/sam/bowtie2_summary/${histName}_bowtie2_spikeIn.txt
seqDepthDouble=`samtools view -@ $cores -F 0x04 $projPath/alignment/sam/${histName}_bowtie2_spikeIn.sam | wc -l`
seqDepth=$((seqDepthDouble/2))
echo $seqDepth >$projPath/alignment/sam/bowtie2_summary/${histName}_bowtie2_spikeIn.seqDepth
##############
echo "$histName Convert Sam to Bam and Bed"
echo Job started at `date`
## Filter and keep the mapped read pairs
samtools view -bS -F 0x04 $projPath/alignment/sam/${histName}_bowtie2.sam >$projPath/alignment/bam/${histName}_bowtie2.mapped.bam
## Convert into bed file format
bedtools bamtobed -i $projPath/alignment/bam/${histName}_bowtie2.mapped.bam -bedpe >$projPath/alignment/bed/${histName}_bowtie2.bed
## Keep the read pairs that are on the same chromosome and fragment length less than 1000bp.
awk '$1==$4 && $6-$2 < 1000 {print $0}' $projPath/alignment/bed/${histName}_bowtie2.bed >$projPath/alignment/bed/${histName}_bowtie2.clean.bed
## Only extract the fragment related columns
cut -f 1,2,6 $projPath/alignment/bed/${histName}_bowtie2.clean.bed | sort -k1,1 -k2,2n -k3,3n >$projPath/alignment/bed/${histName}_bowtie2.fragments.bed
done
multiqc result
###bowtie2 result####
###To see the alignment result########
####multiqc#####
multiqc $projPath/alignment/sam/*bowtie2.sam $projPath/alignment/sam/
Spike-in calibration
fragments
###############
for histName in H1_HU H2_HU H3_HU_IgG H4_HU H5_HU H6_HU_IgG H7_HU N1 N2 N1_HU N2_HU N3_HU_IgG N4_HU N5 N5_HU N6 N6_HU_IgG N7_HU N7_PC N8_NC H_HU_IgG N_HU_IgG
do
#Spike-in calibration
echo "$histName Normalized bedgraph"
echo Job started at `date`
if [[ "$seqDepth" -gt "1" ]]; then
seqDepthDouble=`samtools view -@ $cores -F 0x04 $projPath/alignment/sam/${histName}_bowtie2_spikeIn.sam | wc -l`
seqDepth=$((seqDepthDouble/2))
scale_factor=`echo "10000 / $seqDepth" | bc -l`
##### echo "Scaling factor for $histName is: $scale_factor!"#####
bedtools genomecov -bg -scale $scale_factor -i $projPath/alignment/bed/${histName}_bowtie2.fragments.bed -g $chromSize &>$projPath/alignment/bedgraph/${histName}_bowtie2.fragments.normalized.bedgraph
fi
done
PeakCalling
获得两个结果,一个是与「IgG」进行对比后,去除了IgG的背景【
{histName}seacr_top0.01.peaks】。
不同分组最好是用不同的IgG control, 在这里我是只用了N8_NC我们的IgG control, 由于篇幅有限没有放上其他分组的以不同IgG control为control的代码。
「如果数据正常的条件下,control.peaks 是至少能 calling 到 几M 的peak 。」
#differnt group have different IgG (histControl)
for histName in H1_HU H2_HU H3_HU_IgG H4_HU H5_HU H6_HU_IgG H7_HU N1 N2 N1_HU N2_HU N3_HU_IgG N4_HU N5 N5_HU N6 N6_HU_IgG N7_HU N7_PC N8_NC H_HU_IgG N_HU_IgG
do
echo"$histName"
#Peak calling with SEACR.sh with both control and top 0.01
echo "$histName peak calling"
echo Job started at `date`
bash $seacr $projPath/alignment/bedgraph/${histName}_bowtie2.fragments.normalized.bedgraph \
$projPath/alignment/bedgraph/${histControl}_bowtie2.fragments.normalized.bedgraph \
non stringent $projPath/peakCalling/SEACR/${histName}_seacr_control.peaks
bash $seacr $projPath/alignment/bedgraph/${histName}_bowtie2.fragments.normalized.bedgraph 0.01 non stringent $projPath/peakCalling/SEACR/${histName}_seacr_top0.01.peaks
done
Computematrix
「这部分算是数据可视化部分了,plotHeatmap运行的时候如果用的是全部matrix记得要留足内存,要不容易失败。」
for histName in H1_HU H2_HU H3_HU_IgG H4_HU H5_HU H6_HU_IgG H7_HU N1 N2 N1_HU N2_HU N3_HU_IgG N4_HU N5 N5_HU N6 N6_HU_IgG N7_HU N7_PC N8_NC H_HU_IgG N_HU_IgG
do
samtools sort -o $projPath/alignment/bam/${histName}.sorted.bam $projPath/alignment/bam/${histName}_bowtie2.mapped.bam
samtools index $projPath/alignment/bam/${histName}.sorted.bam
bamCoverage -b $projPath/alignment/bam/${histName}.sorted.bam -o $projPath/alignment/bigwig/${histName}_raw.bw
########computeMatrix#######
computeMatrix scale-regions -S $projPath/alignment/bigwig/${histName}_raw.bw \
-R /home/cheny5/han00433/project/GRCh38_noalt_as/hg38_RefSeq.bed \
--beforeRegionStartLength 3000 \
--regionBodyLength 5000 \
--afterRegionStartLength 3000 \
--skipZeros -o $projPath/peakCalling/${histName}_matrix_gene.mat.gz -p $cores
plotHeatmap -m $projPath/peakCalling/${histName}_matrix_gene.mat.gz -out $projPath/peakCalling/${histName}_gene.png --sortUsing sum
done
作为例子在此只把postive control展示出来。
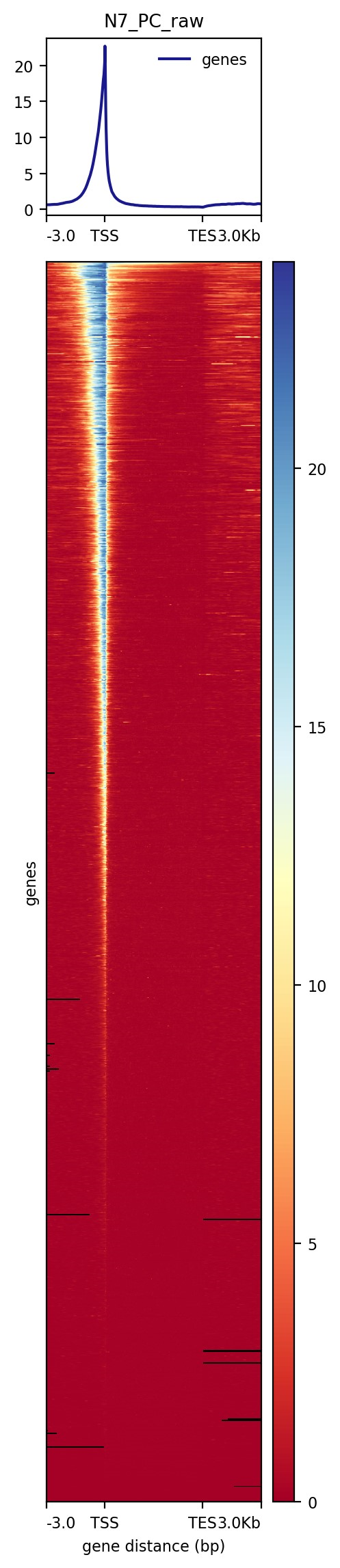
Computematrix—2
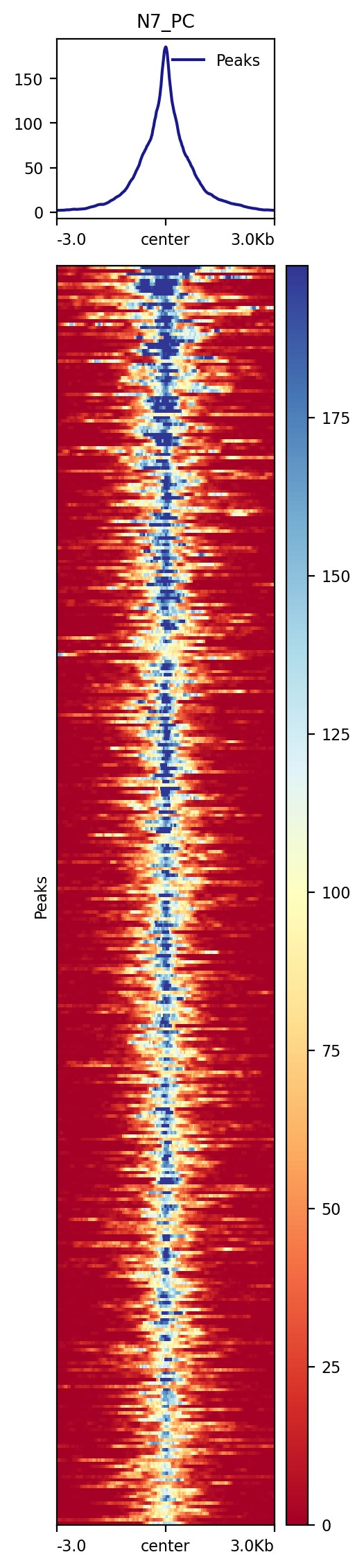
「CUT&Tag peaks 区域热图」
使用从 SEACR 返回的信号区域的中点来对齐热图中的信号。
SEACR 输出的 「第六列是 chr:start-end」 形式的文件,表示该区域信号最大的区域的开始和结尾位置。
我们首先生成一个 「包含 6 列中的中点信息的新 bed 文件」,并使用 「plotHeatmap」 来实现热图可视化。
for histName in H1_HU H2_HU H3_HU_IgG H4_HU H5_HU H6_HU_IgG H7_HU N1 N2 N1_HU N2_HU N3_HU_IgG N4_HU N5 N5_HU N6 N6_HU_IgG N7_HU N7_PC N8_NC H_HU_IgG N_HU_IgG
do
awk '{split($6, summit, ":"); split(summit[2], region, "-"); print summit[1]"\t"region[1]"\t"region[2]}' $projPath/peakCalling/SEACR/${histName}_seacr_control.peaks.stringent.bed >$projPath/peakCalling/SEACR/${histName}_seacr_control.peaks.summitRegion.bed
computeMatrix reference-point -S $projPath/alignment/bigwig/${histName}_raw.bw \
-R $projPath/peakCalling/SEACR/${histName}_seacr_control.peaks.summitRegion.bed \
--skipZeros -o $projPath/peakCalling/${histName}_SEACR.mat.gz -p $cores -a 3000 -b 3000 --referencePoint center
plotHeatmap -m $projPath/peakCalling/${histName}_SEACR.mat.gz -out $projPath/peakCalling/${histName}_SEACR_heatmap.png --sortUsing sum --startLabel "Peak Start" -\
-endLabel "Peak End" --xAxisLabel "" --regionsLabel "Peaks" --samplesLabel "${histName}"
done
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-10-26,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号