作者仅提供了fpkm格式表达量矩阵的转录组测序数据集该如何重新分析呢

作者仅提供了fpkm格式表达量矩阵的转录组测序数据集该如何重新分析呢

生信技能树
发布于 2024-06-08 08:26:27
发布于 2024-06-08 08:26:27
一个2021的糖尿病转录组文章:《Altered human alveolar bone gene expression in type 2 diabetes—A cross-sectional study》,在线链接是:https://onlinelibrary.wiley.com/doi/abs/10.1111/jre.12947
研究者们在GEO数据库是有数据分享:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE182923
- RNA samples were further purified using ribosomal RNA depletion technique and processed for RNA sequencing and analysis.
- Expression levels for mRNAs were performed by calculating FPKM ([total_exon_fragments/mapped reads (millions) × exon length (kB)]),
- 最后是差异分析后确定统计学阈值:differentially expressed mRNAs were selected with log2 (fold change) >1 or log2 (fold change) ≤1 and with a parametric F test comparing nested linear models.
可以看到的是作者给出来的是57.5 Mb 的矩阵文件 :
GSE182923_genes_fpkm_expression.txt.gz
转录组测序数据的表达量矩阵可以有多种格式,每种格式都有其特定的用途和优势。以下是一些常见的格式:
- 计数矩阵(Count Matrix):
- 这是最基本的格式,通常由比对到参考基因组的读段生成。
- 每一行代表一个基因或转录本,每一列代表一个样本。
- 单元格中的值表示该基因在该样本中的读段计数。
- FPKM/FPKM-UQ(每千个碱基每百万片段的比率/未量化的FPKM):
- FPKM是标准化的表达量指标,考虑了基因长度和测序深度。
- FPKM-UQ是未量化的FPKM,它没有经过标准化处理,通常用于避免引入人为的表达量变化。
- TPM(每千个转录本每百万片段的比率):
- TPM是另一种标准化的表达量指标,它考虑了样本中的总转录本数。
- TPM使得不同样本间的基因表达量可比。
- CPM(每百万计数的比率):
- CPM是一种简单的标准化方法,将计数除以样本的总计数乘以百万。
- 它用于归一化数据,使得不同样本间的表达量可比。
- RSEM/Cufflinks输出:
- RSEM(RNA-Seq by Expectation-Maximization)和Cufflinks是两种软件工具,它们提供了一种估计基因和转录本表达量的方法。
- 输出通常包括每个基因的估计表达量(如FPKM)、表达量的不确定性和统计评估。
- Salmon输出:
- Salmon是一种用于RNA-Seq数据的无需比对的定量工具,它使用轻量级比对和EM算法来估计表达量。
- 输出通常包括每个转录本的TPM和预期计数(expected count)。
我们通常是针对转录组测序使用DESeq2/edgeR进行差异分析,而DESeq2/edgeR要求的输入数据是计数矩阵(raw Count Matrix)格式,作者并没有提供,而且我们不可能依据作者提供的FPKM矩阵去反推出来原始的计数矩阵(raw Count Matrix)。
这里我们推荐:https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE182923
而且这个geo2r网页工具还贴心的给出来了代码,如下所示:
# Version info: R 4.2.2, Biobase 2.58.0, GEOquery 2.66.0, limma 3.54.0
################################################################
# Data plots for selected GEO samples
# load counts table from GEO
urld <- "https://www.ncbi.nlm.nih.gov/geo/download/?format=file&type=rnaseq_counts"
path <- paste(urld, "acc=GSE182923", "file=GSE182923_raw_counts_GRCh38.p13_NCBI.tsv.gz", sep="&");
tbl <- as.matrix(data.table::fread(path, header=T, colClasses="integer"), rownames=1)
# pre-filter low count genes
# keep genes with at least 2 counts > 10
keep <- rowSums( tbl >= 10 ) >= 2
tbl <- tbl[keep, ]
# log transform raw counts
# instead of raw counts can display vst(as.matrix(tbl)) i.e. variance stabilized counts
dat <- log10(tbl + 1)
# box-and-whisker plot
par(mar=c(7,4,2,1))
boxplot(dat, boxwex=0.7, notch=T, main="GSE182923", ylab="lg(cnt + 1)", outline=F, las=2)
# UMAP plot (dimensionality reduction)
library(umap)
dat <- dat[!duplicated(dat), ] # first remove duplicates
ump <- umap(t(dat), n_neighbors = 5, random_state = 123)
plot(ump$layout, main="GSE182923 UMAP plot, nbrs =5", xlab="", ylab="", pch=20, cex=1.5)
library(car)
pointLabel(ump$layout, labels = rownames(ump$layout), method="SANN", cex=0.6)
值得注意的是表达量矩阵并不是每个样品会被纳入
可以看到的是作者虽然是 Cross-sectional, no replicates, 10 healthy, 8 diabetic,但是geo2r仅仅是纳入了4个疾病和7个对照哦,我推测应该是这个数据集的测序质量很差,有一些样品不满足前面的转录组定量要求就被暴力删除了,其实也是合理的选择样品 :
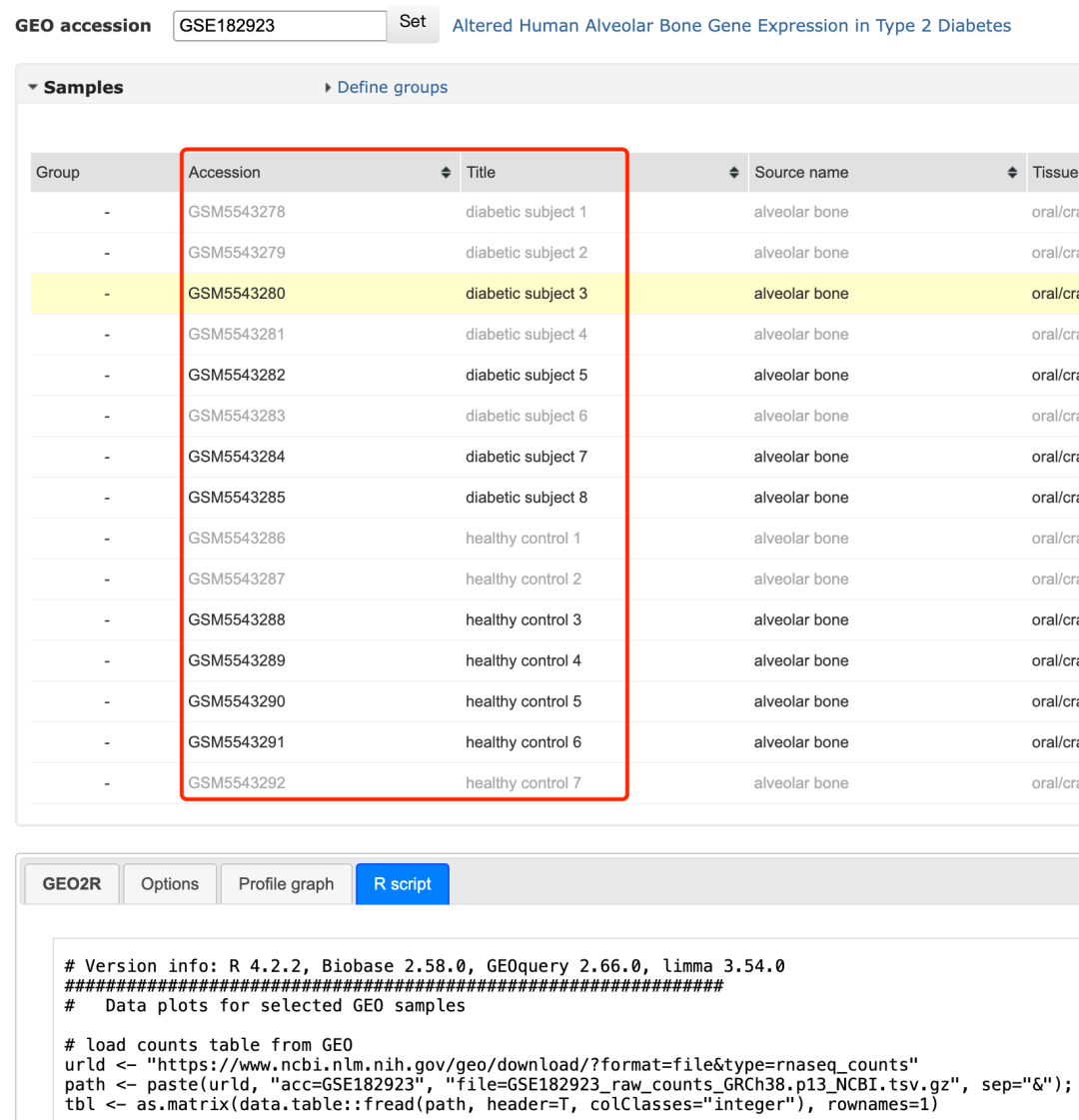
不满足前面的转录组定量要求就被暴力删除了
当然了,就算是我们拿到了DESeq2/edgeR要求的输入数据是计数矩阵(raw Count Matrix)格式的文件,做后面的差异分析也很难,因为文章自己就一个很垃圾的差异分析结果,如下所示:
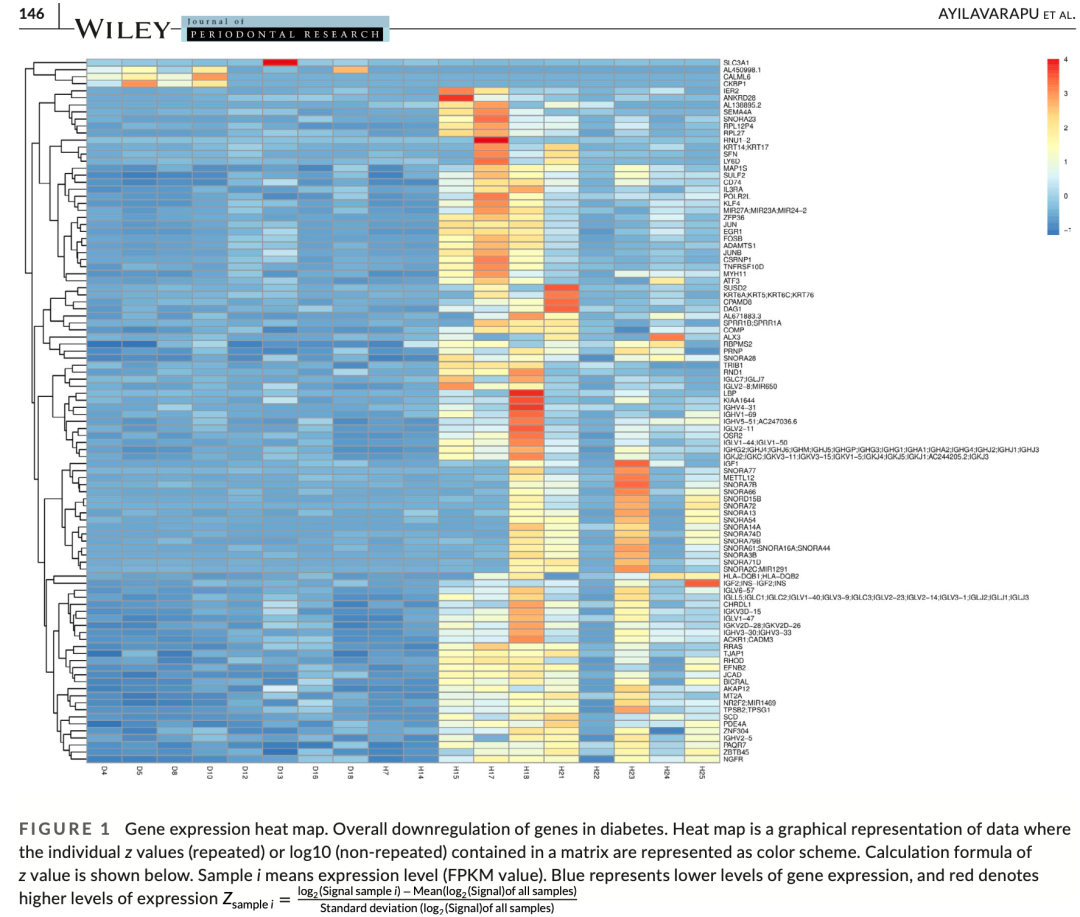
很垃圾的差异分析结果
GEO数据库的任意转录组测序数据集均可获得count矩阵
虽然说上面的案例(糖尿病数据集GSE182923)是因为作者自己的原因导致我们虽然是获得count矩阵但是差异分析结果也丑爆了。但是这个解决方案是 通用的, 理论上GEO数据库的任意转录组测序数据集均可获得count矩阵。比如这个GSE148241-先兆子痫-数据集,是 placentae from 9 patients with early-onset severe preeclampsia (EOSPE) and 32 normal controls, 同样的方式获取count矩阵和分组信息 :
# load counts table from GEO
urld <- "https://www.ncbi.nlm.nih.gov/geo/download/?format=file&type=rnaseq_counts"
path <- paste(urld, "acc=GSE148241", "file=GSE148241_raw_counts_GRCh38.p13_NCBI.tsv.gz", sep="&");
path
tbl <- as.matrix(data.table::fread(path, header=T, colClasses="integer"), rownames=1)
# data<-data.table::fread("matrix.txt",
# data.table = F)
dim(tbl)
mat=as.data.frame(tbl)
library(AnnoProbe)
library(GEOquery)
gset = getGEO("GSE148241", destdir = '.', getGPL = F)
pd = pData(gset[[1]])
table(group_list)
group_list=ifelse(grepl('Normal',pd$source_name_ch1),
'control','case' )
就可以常规的差异分析,如下所示的火山图和热图:
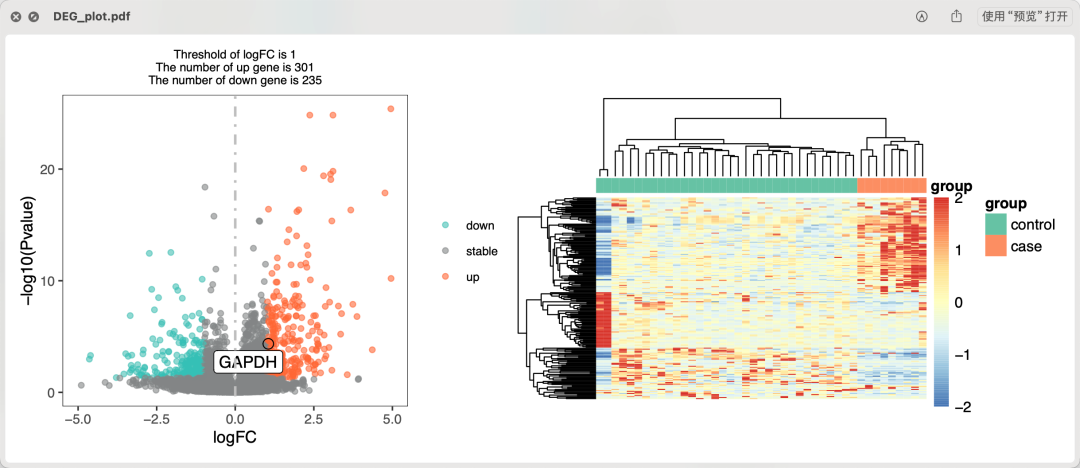
火山图和热图
可以看到有两个样品是离群点, 其实这个GSE148241-先兆子痫-数据集页面也指出来了,但是数据集配套的文献并没有关心这个差异分析结果,反而是做了一个wgcna分析。如果你恰好是先兆子痫研究方向, 就可以把这个数据集更加细致的解读和挖掘一下,未必不是一个课题哦!
写在文末
如果你确实觉得我的教程对你的科研课题有帮助,让你茅塞顿开,或者说你的课题大量使用我的技能,烦请日后在发表自己的成果的时候,加上一个简短的致谢,如下所示:
We thank Dr.Jianming Zeng(University of Macau), and all the members of his bioinformatics team, biotrainee, for generously sharing their experience and codes.
十年后我环游世界各地的高校以及科研院所(当然包括中国大陆)的时候,如果有这样的情谊,我会优先见你。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-06-05,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号