GEO数据分析流程之芯片1补充


生新技能树学习笔记
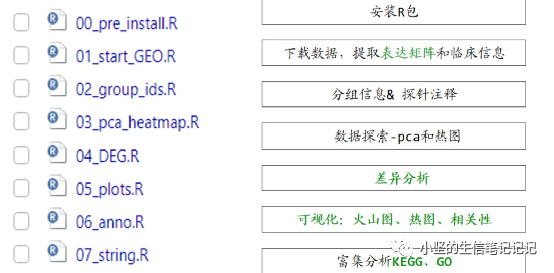
代码分析流程
第一步:安装R包
options("repos"="https://mirrors.ustc.edu.cn/CRAN/")if(!require("BiocManager")) install.packages("BiocManager",update = F,ask = F)options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")cran_packages <- c('tidyr', 'tibble', 'dplyr', 'stringr', 'ggplot2', 'ggpubr', 'factoextra', 'FactoMineR', 'devtools', 'cowplot', 'patchwork', 'basetheme', 'paletteer', 'AnnoProbe', 'ggthemes', 'VennDiagram', 'tinyarray') Biocductor_packages <- c('GEOquery', 'hgu133plus2.db', 'ggnewscale', "limma", "impute", "GSEABase", "GSVA", "clusterProfiler", "org.Hs.eg.db", "preprocessCore", "enrichplot") for (pkg in cran_packages){ if (! require(pkg,character.only=T) ) { install.packages(pkg,ask = F,update = F) require(pkg,character.only=T) }} for (pkg in Biocductor_packages){ if (! require(pkg,character.only=T) ) { BiocManager::install(pkg,ask = F,update = F) require(pkg,character.only=T) }} |
|---|
第二步 检查安装是否成功
#前面的所有提示和报错都先不要管。主要看这里for (pkg in c(Biocductor_packages,cran_packages)){ require(pkg,character.only=T) } |
|---|
第三步 确定数据研究方向、物种、数据类型(一定要是芯片,标注array)

样品信息

平台
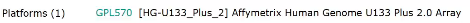
点开Matrix

当这里的大小为M,证明数据完整,如果是K,说明数据可能缺失
第四步 运行代码 下载数据
#数据下载rm(list = ls())#清空数据library(GEOquery)#加载包#先去网页确定是否是表达芯片数据,不是的话不能用本流程。gse_number = "GSE56649"eSet <- getGEO(gse_number, destdir = '.', getGPL = F)#下载并读取class(eSet)#列表length(eSet)#列表长度eSet = eSet[[1]]#提取列表#(1)提取表达矩阵expexp <- exprs(eSet) #提取dim(exp)exp[1:4,1:4] #正常范围在0-20之间#检查矩阵是否正常,如果是空的就会报错,空的和有负值的、有异常值的矩阵需要处理原始数据。#如果表达矩阵为空,大多数是转录组数据,不能用这个流程(后面另讲)。#自行判断是否需要logexp = log2(exp+1) #这一行下载的数据如果取过log,就不要运行。如果在几百,需要取log。如果在0-20,不用取log。为了避免0值,exp+1.甲基化+0.0001,为了保证取log后不会有负无穷。boxplot(exp)#(2)提取临床信息pd <- pData(eSet) #pd文件是样本信息#(3)让exp列名与pd的行名顺序完全一致p = identical(rownames(pd),colnames(exp));p #exp是表达矩阵,identical表示判断是否一致if(!p) exp = exp[,match(rownames(pd),colnames(exp))] #match#(4)提取芯片平台编号gpl_number <- eSet@annotation;gpl_number#@和$都表示提取子集,可以通过补齐判断,或者用str().save(gse_number,pd,exp,gpl_number,file = "step1output.Rdata") |
|---|
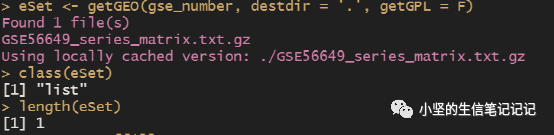
提取的转化后的eSet格式为ExpressionSet 可以被Biobase(R包)提取。(狭义的对象,是被R包的作者定义的)。

这里可以显示下数据有多少行,小于2万行,可能数据不完整

验证数据是否正常
boxplot(exp)
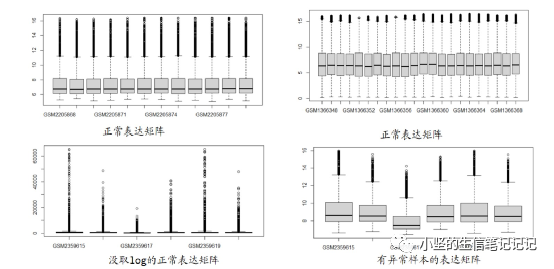
异常值的处理办法 1.删掉异常样本
2.exp=limma::normalizeBetweenArrays(exp)
关于表达矩阵里的负值
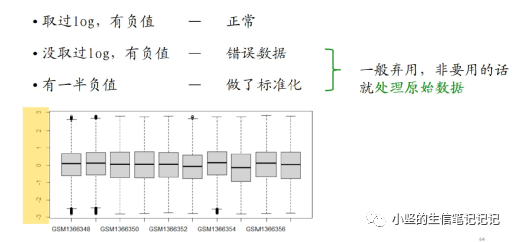
有一半的负值可以做热图,不做差异分析。
关于原始数据

下载的加速函数,可以替代最开始的下载数据函数
rm(list = ls())library(GEOquery)library(AnnoProbe)#先去网页确定是否是表达芯片数据,不是的话不能用本流程。gse_number = "GSE56649"eSet = geoChina(gse_number) |
|---|
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-06-27,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号