R包安装总是出错?这份汇总帮你一键解决!


为了方便用户高效开展生物信息学研究,天意云生信技术团队已在服务器环境中预装了上千款常用的生信软件及R语言软件包。这极大地简化了标准分析流程的部署。
然而,特定的研究课题往往需要依赖一些未包含在预装列表中的专用工具或软件包。因此,不可避免地,在研究过程中可能会遇到需要自行安装特定R包的情况。
本篇推文小编根据长期观察和用户反馈,特意梳理并汇总了在服务器上安装R包过程中最常遇到的问题及其可能的解决思路。希望能为大家提供一份实用的参考指南,助力各位在服务器上顺利、高效地开展数据分析和研究工作。
当然,如果在安装过程中遇到任何难以解决的问题,请不必犹豫,随时联系我们的技术支持团队寻求帮助。
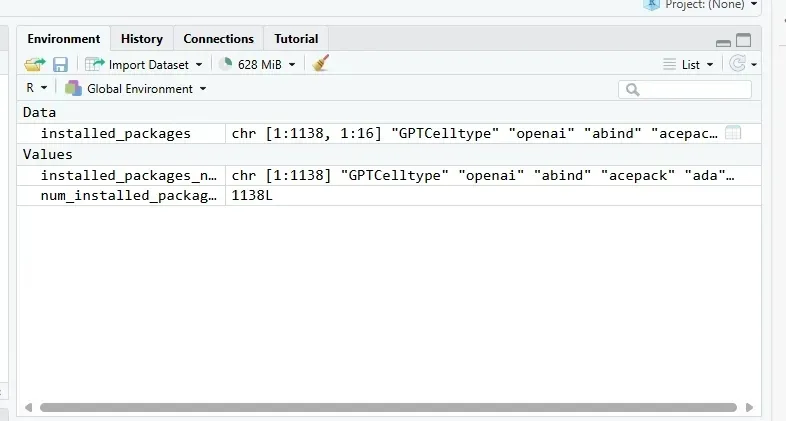
安装R包的常规流程
1、使用install.packages()函数
这是最常用的在线安装方法,适用于CRAN上的大部分R包。用户只需在R控制台输入如下命令:
install.packages("package_name")如果需要同时安装多个包,可以使用:
install.packages(c("package_name1", "package_name2"))此外,还可以通过设置repos参数指定不同的镜像源,例如:
install.packages("package_name", repos = "https://cran.cnn.com")这种方法简单直接,但有时会因网络问题导致下载速度较慢。
2、使用BiocManager::install()函数
对于Bioconductor项目中的包,推荐使用BiocManager包进行安装。
例如:
BiocManager::install("package_name")此方法适用于生物信息学相关的R包,如ggplot2等。
3、从GitHub安装
如果需要安装尚未发布到CRAN或Bioconductor的包,可以通过devtools或remotes包实现。例如:
devtools::install_github("username/package_name")或者:
remotes::install_github("username/package_name")这种方法适合开发者直接从源码安装最新版本的包。
4、本地下载安装
用户可以从CRAN或GitHub等平台下载R包的压缩文件,然后通过以下命令进行本地安装:
install.packages("path/to/package.zip", repos = NULL, type = "source")这种方法适合网络环境较差或需要特定版本的用户。
用conda安装包
conda具备环境隔离与依赖管理的能力,有时候会解决版本不兼容、依赖缺失等问题。我们的服务器已经配置好了conda,一般不低于4个conda环境,分别对应不同的功能。用户也可以自行按需创建。
一些“顽固”R包的安装方法
1、安装scMetabolism
官方教程:https://github.com/wu-yc/scMetabolism
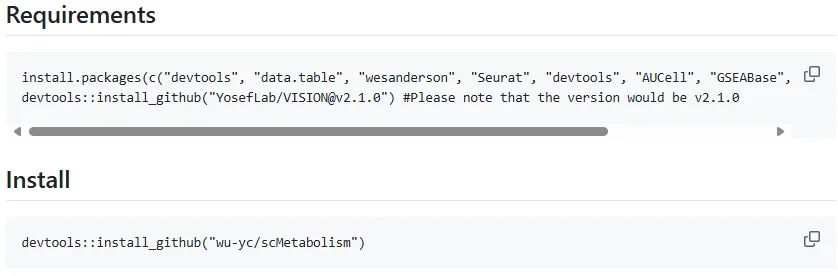
用户在安装VISION@v2.1.0的时候大概率会缺失‘loe’, ‘plumber’这两个包。
使用conda安装:
conda install conda-forge::r-loe
conda install conda-forge::r-plumber
然后:
devtools::install_github("YosefLab/VISION@v2.1.0")
devtools::install_github("wu-yc/scMetabolism")
2、github TOKEN的问题
从github上下载包的时候可能会遇到权限问题。提示的方式有很多种,这里不再罗列报错内容。
解决办法:
(1)创建 GitHub Token:
- 访问 GitHub 网站:GitHub Personal Access Token 创建页面
- 点击“Generate new token”生成一个新的访问令牌,选择适当的权限(如 repo,read:user 等)。
- 生成后复制该 Token。
(2)在 R 中使用该 Token
install.packages("gitcreds")
library(gitcreds)
gitcreds::gitcreds_set()
(3)重新尝试安装
完成上述步骤后,重新运行安装命令,例如:
devtools::install_github("ChiLiubio/microeco")
3、安装cmdstanr
当我尝试使用 cmdstanr 在 R 中编译 Stan 模型时,出现错误。make 命令在执行时失败了,具体原因是它无法找到一个名为 make/command 的文件或目标。

重新安装:
git clone https://github.com/stan-dev/cmdstan.git --recursive
cd cmdstan
make build
在使用make编译cmdstan时,指定标准编辑器:make build CXX=g++。
在 R 中明确指定 CmdStan 的路径:
library(cmdstanr)
set_cmdstan_path("/path/to/cmdstan")
model <- cmdstan_model('/path/to/dp.log.joint_model.sigma.stan',
cpp_options = list(stan_threads = TRUE))
4、安装beyondcell
conda、mamba都安装不了,因为其他软件的版本问题,这里就需要常规的手动安装的方式。
https://github.com/cnio-bu/beyondcell
(1)手动下载最新版本的beyondcell
https://github.com/cnio-bu/beyondcell/archive/refs/tags/v2.2.2.tar.gz
(2)在R环境中安装
install.packages("/mnt/data/tool/beyondcell_v2.tar.gz",repos=NULL)
5、安装harmony
单细胞转录组分析中多样本整合的工具包harmony,有用户按照常规方法安装失败。
最简单的方法:
conda install --no-channel-priority conda-forge::r-harmony
这个命令会忽略仓库的优先级,直接从 conda-forge 安装包。
6、suerat降级
我们的服务器上配置好的是V5,但是有的用户,使用的方法和教程是V4的,所以需要进行降级操作。
#先关掉R,再打开,清理掉缓存变量
remove.packages(c("Seurat","SeuratObject"))
install.packages('Seurat', repos = c('https://satijalab.r-universe.dev'))
这个镜像安装的是suerat4.4的版本
7、安装sodium
sodium 是一个 R 包,它提供了用于加密、哈希和数字签名等功能的工具,这个包基于 Libsodium 库,而 Libsodium 是一个用于加密的 C 库。
安装方法:
conda install conda-forge::libsodium
install.packages("sodium")
Tips:大家如果在服务器端安装R包,一定要特别注意R包的安装路径是否与RStudio中调用这些包的路径一致!
这是一个非常容易被忽略、但又极其关键的细节。尤其是在服务器环境中,多个用户之间也可能使用不同的库路径(library path)。你可以用.libPaths()查看当前的包搜索路径,也可以通过设置.libPaths("/your/path")来显式指定安装和加载包的位置。
总结
以上就是用户经常咨询的R包的安装方法。细心的朋友可能已经发现,安装那些稍微复杂一点的R包,其实不过是常规方法的灵活排列组合。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-04-09,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号