AI+Drug 文献速递 | HADDOCK3:构建自定义流程,助力生物分子复合物建模研究

AI+Drug 文献速递 | HADDOCK3:构建自定义流程,助力生物分子复合物建模研究

MindDance
发布于 2026-01-08 12:45:10
发布于 2026-01-08 12:45:10
1. HADDOCK3: A modular and versatile platform for integrative modelling of biomolecular complexes
期刊: biorxiv 链接: https://doi.org/10.1101/2025.04.30.651432 代码: https://github.com/haddocking/haddock3
简介: HADDOCK3是一款用于生物分子复合物整合建模的开源软件,其创新性地采用模块化设计,可灵活构建自定义工作流程。它将原有的固定流程分解为独立模块,涵盖拓扑、采样、细化、评分和分析等类别。在实验方面,通过多个案例展示了其在多种场景下的应用效果。如在抗体-抗原复合物建模中,可利用多组约束确定正确结合界面;在蛋白质-聚糖对接时,添加聚类步骤提升了对接结果。使用的数据集包括PDB数据库等。结果表明,HADDOCK3能有效解决之前版本无法处理的问题,为结构生物学研究提供了有力工具。

2. MolMole: Molecule Mining from Scientific Literature
期刊: arxiv 链接: https://arxiv.org/abs/2505.03777v1 代码: https://lgai-ddu.github.io/molmole/
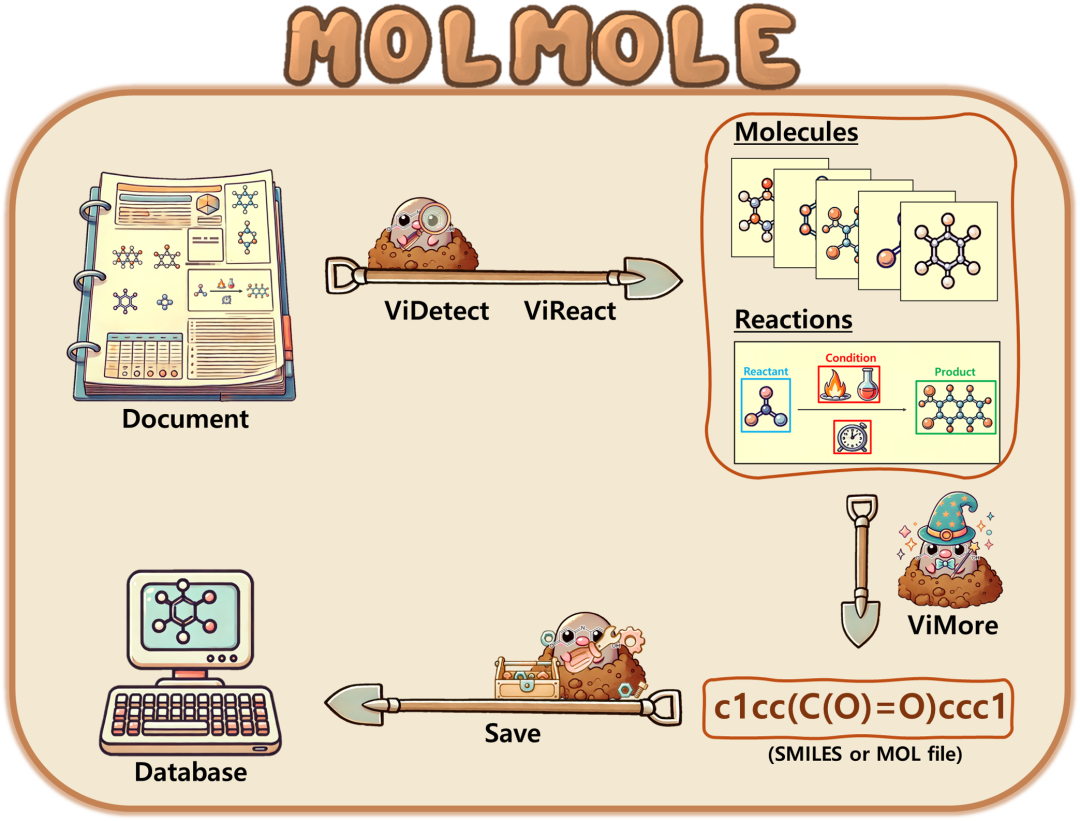
简介: MolMole是一种基于视觉的深度学习框架,能从科学文献中提取分子结构和反应数据,其创新点在于将分子检测、反应图解析和光化学结构识别集成于同一流程,并构建新的基准数据集和评估指标。它通过ViDetect、ViReact和ViMore三个模块处理文档图像,分别实现分子结构检测、反应图解析和分子图像格式转换。实验使用自定义的550页文档数据集,包含3897个分子结构和1022个反应。结果显示,MolMole在分子检测和OCSR综合性能上,F1分数分别达到89.1%和86.8%,在反应图解析上F1分数分别为98.0%和97.0%,均优于现有工具包。该框架为化学数据提取提供了高效准确的方法。
3. Fragment-Masked Molecular Optimization
期刊: arxiv 链接: https://arxiv.org/abs/2408.09106v2 代码: https://anonymous.4open.science/r/FMOP-98C2
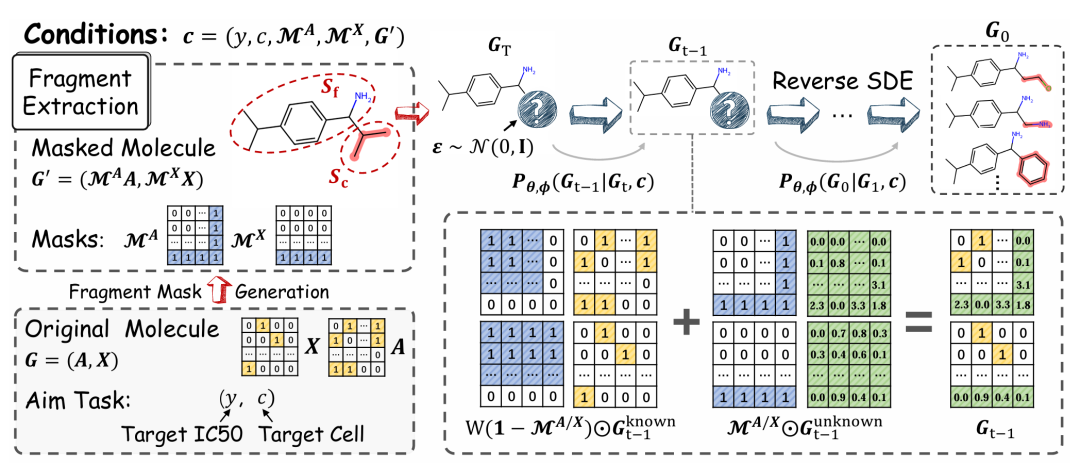
简介: 论文提出基于表型药物发现(PDD)的片段掩码分子优化方法(FMOP),旨在解决PDD中分子优化难题,这是其创新之处。该方法利用无回归扩散模型,结合分子片段掩码和规则约束,在无需训练的情况下优化分子掩码区域。实验使用QM9和GDSCv2数据集,在GDSCv2数据集的945个细胞系上进行优化实验。结果显示,FMOP的计算机模拟优化成功率达94.4%,平均疗效提高5.3%,且在与其他五种基线模型对比中表现最佳。此外,消融实验和可视化分析验证了该方法的有效性和稳健性,为PDD分子优化提供了新途径。
4. Sparsity is All You Need: Rethinking Biological Pathway-Informed Approaches in Deep Learning
期刊: arxiv 链接: https://arxiv.org/abs/2505.04300v1 代码: https://github.com/compbiomed-unito/Pathway_Randomization
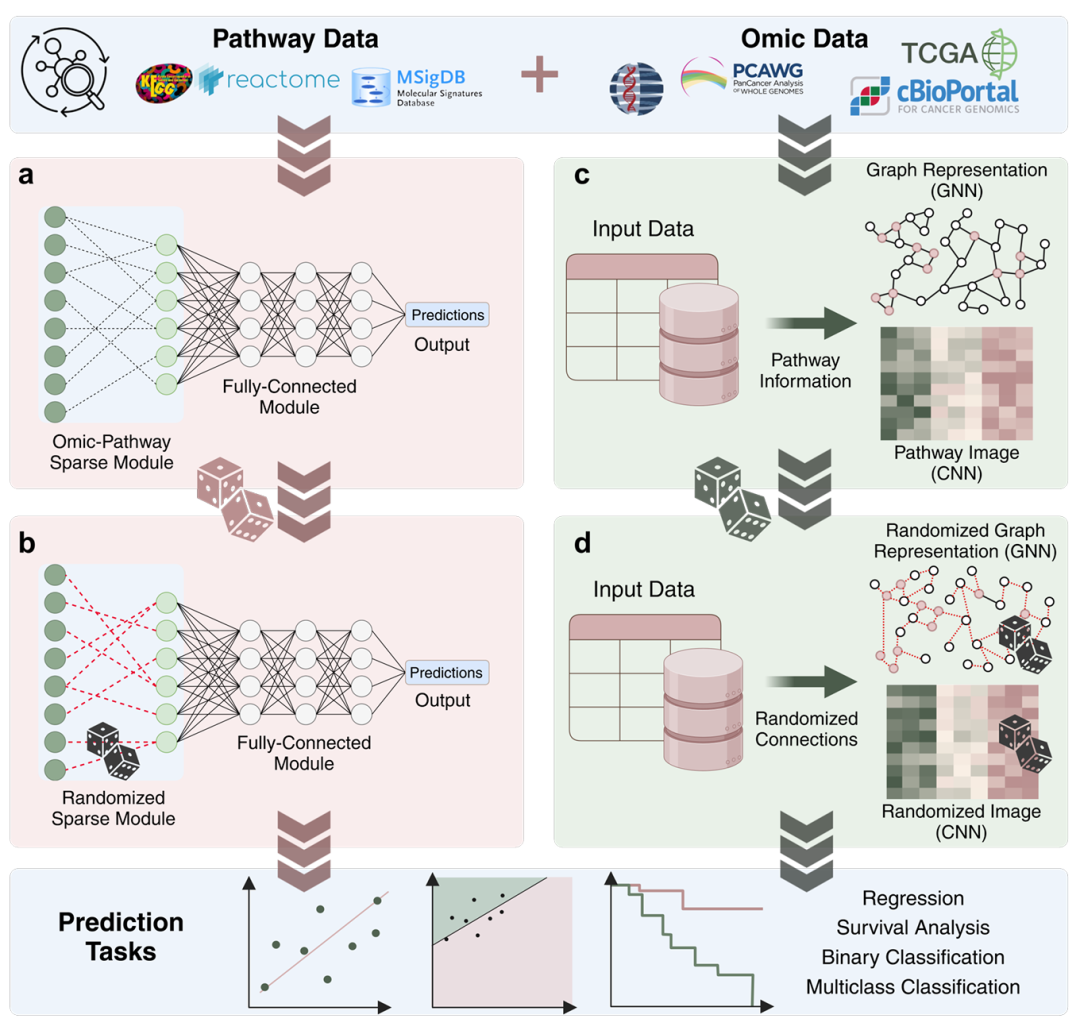
简介: 该论文重新思考了深度学习中生物通路信息方法,发现通路整合的优势可能源于其引入的稀疏性而非生物学相关性,并提出新的比较方法。研究人员通过随机化实验,对比基于通路信息的深度学习模型和使用随机信息但保留稀疏性的模型性能。实验使用多个包含基因表达等数据的数据集,涉及多种预测任务。结果显示,随机化模型在多项指标上表现与原模型相当甚至更优,且在某些模型中,随机化版本显著超越原模型,同时发现生物通路诱导的稀疏性并非最优。该研究为生物先验整合到机器学习框架的研究提供了新方向,促使研究人员重新审视相关模型的设计重点。
5. GRAPE: Heterogeneous Graph Representation Learning for Genetic Perturbation with Coding and Non-Coding Biotype
期刊: arxiv 链接: https://arxiv.org/abs/2505.03853v1 代码: https://anonymous.4open.science/r/GRAPE-EB39
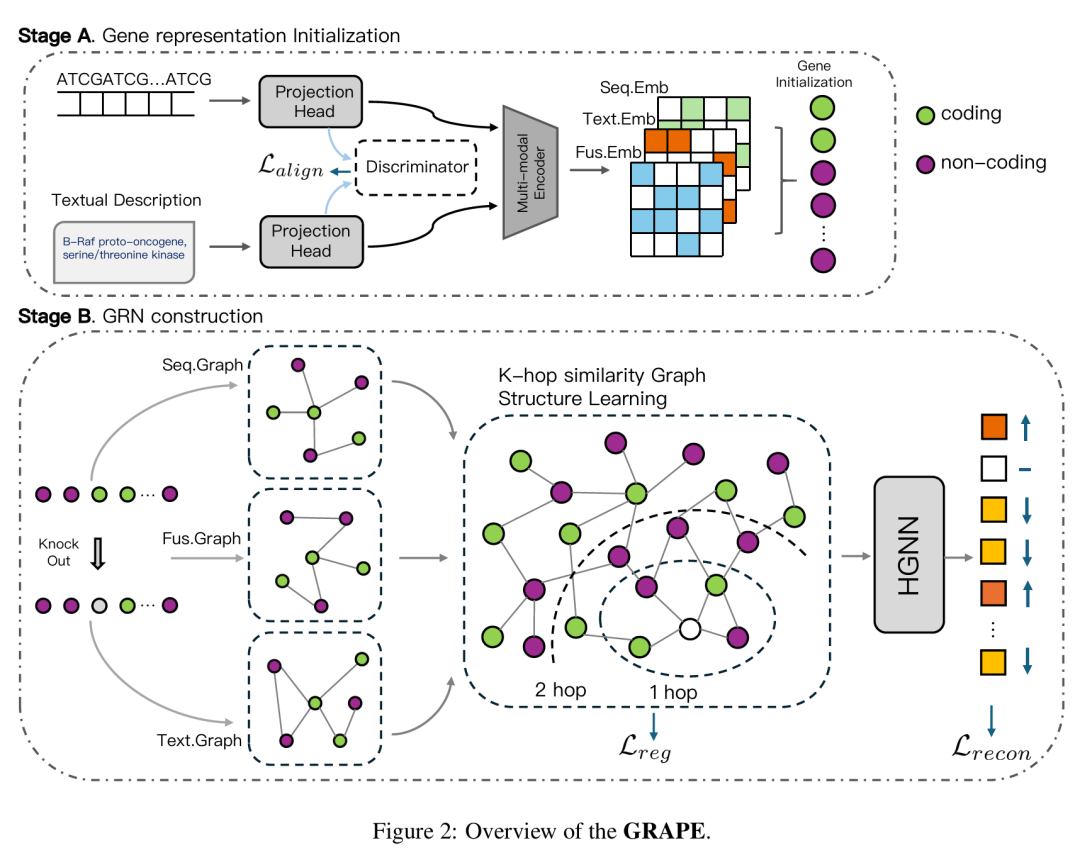
简介: GRAPE模型创新性地将基因生物型信息融入基因调控网络建模,用于预测基因扰动,提升了预测准确性。该模型利用预训练的大语言模型和DNA序列模型提取基因特征,构建异质图并通过图结构学习动态优化基因调控网络。实验采用Adamson和Norman等公开数据集,随机划分训练集和测试集进行实验。结果表明,GRAPE在均方根误差、方向准确率系数等多个评估指标上优于现有方法,且通过实验验证了多模态特征融合、图结构学习和生物型信息引入对模型性能提升的重要性。该研究为基因扰动预测提供了更有效的方法,推动了相关领域的发展。
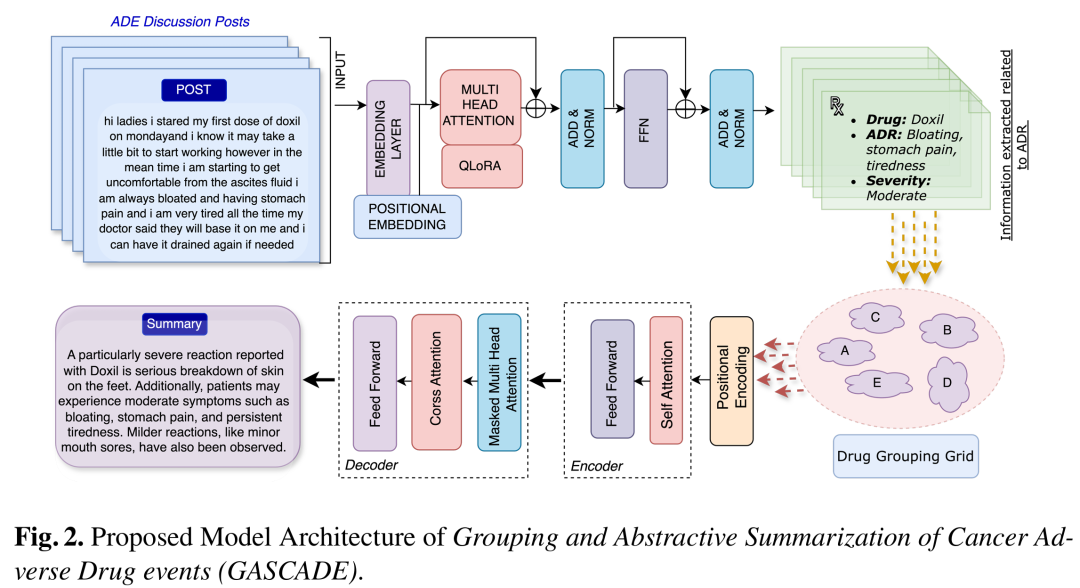
6. GASCADE: Grouped Summarization of Adverse Drug Event for Enhanced Cancer Pharmacovigilance
期刊: arxiv 链接: https://arxiv.org/abs/2505.04284v1 代码: https://github.com/SofeeyaJ/GASCADE_ECIR2025
简介: 论文提出针对癌症药物不良事件(ADE)分组总结的新任务,构建MCADRS数据集并开发GASCADE框架,提升癌症药物警戒能力。GASCADE框架结合大语言模型信息提取能力和T5模型的总结能力,利用QLoRA技术微调模型,通过直接偏好优化(DPO)提升总结质量。实验使用MCADRS数据集,对比多种基线模型,采用自动评估和人工评估相结合的方式。自动评估显示GASCADE在多项指标上表现出色,人工评估表明其生成的总结在临床评估得分、事实召回率等方面优于基线模型。该研究为癌症药物相关决策提供了有力支持,对癌症个性化治疗有重要意义。
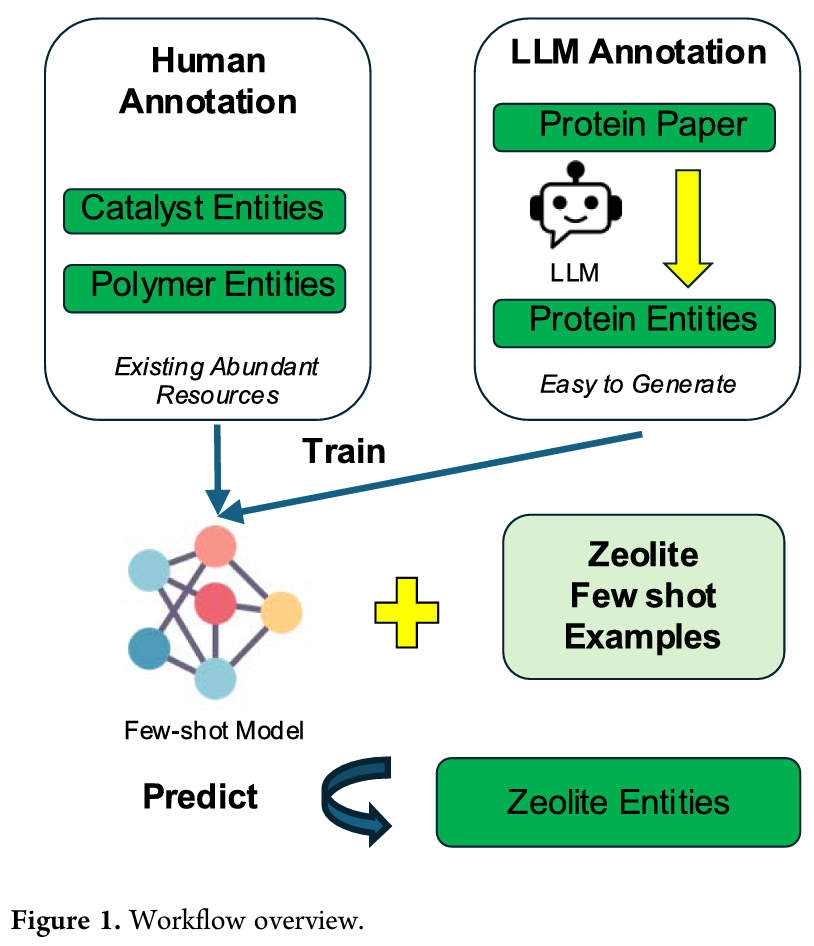
7. Rapid Adaptation of Chemical Named Entity Recognition Using Few-Shot Learning and LLM Distillation
期刊: Journal of Chemical Information and Modeling 链接: https://doi.org/10.1021/acs.jcim.5c00248 代码: https://github.com/nsndimt/ChemSSP
简介: 该论文提出一种少样本化学命名实体识别(NER)模型,能利用少量标注示例快速适应新化学实体提取,创新地将度量学习与大语言模型(LLM)结合,提升模型适应性和效率。模型采用度量学习方法,从高资源化学领域转移实体相似性知识,结合预训练模型和改进的采样算法进行训练。实验使用由六个现有化学NER数据集构建的少样本基准数据集,进行跨域实验。结果显示,模型在仅5个示例/实体类型时就能取得合理性能,且随示例数增加性能提升;与基于LLM的方法相比,该模型在F1得分上优势明显。该研究为化学领域NER提供了更高效的解决方案,推动了相关研究进展。
8. A Foundation Model for Accurate Atomistic Simulations in Drug Design
期刊: chemrxiv 链接: https://doi.org/10.26434/chemrxiv-2025-f1hgn-v3 代码: https://github.com/thomasple/FeNNol https://github.com/TinkerTools/tinker-hp
简介: 文章介绍了FeNNix-Bio1这一用于药物设计中原子模拟的基础模型,它能处理从生物到化学的多种应用,以合成量子数据为基础且可系统改进,突破了传统方法的局限。该模型基于特定的架构设计,利用ωB97M-DFT功能和相关数据集进行训练。实验对水的性质、离子在溶液中的行为等多个方面进行模拟,并与其他模型对比性能。结果表明,FeNNix-Bio1在模拟多种生化问题上表现出色,在计算性能上比MACE-OFF模型更优。该模型为药物设计提供了更准确且计算可行的方法,有望推动药物研发进程。

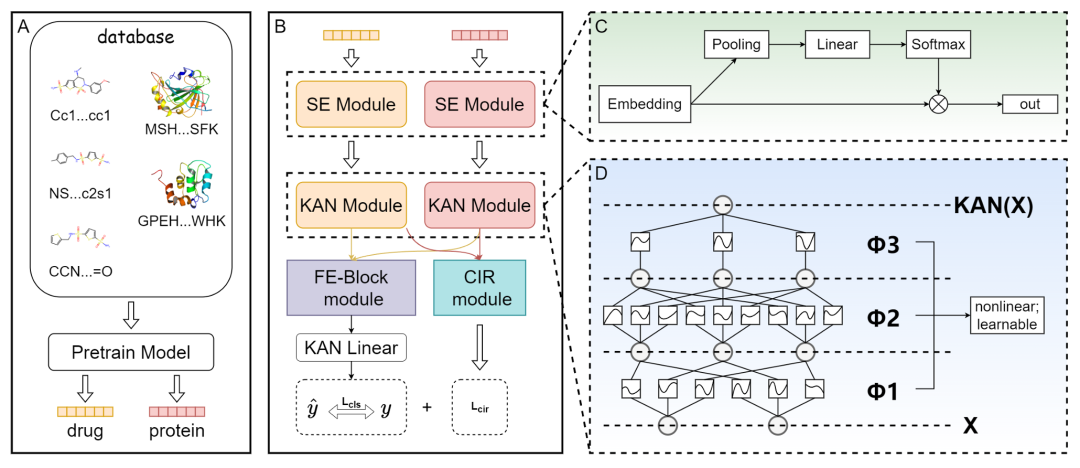
9. DrugKANs: A Paradigm to Enhance Drug-Target Interaction Prediction with KANs
期刊: IEEE Journal of Biomedical and Health Informatics 链接: https://doi.org/10.1109/JBHI.2025.3566931 代码: https://github.com/Excelsior511/DrugKANs
简介: 论文提出DrugKANs模型用于增强药物-靶点相互作用预测,通过整合双塔架构和Kolmogorov-Arnold网络(KAN)技术,提升预测质量和可解释性。模型利用预训练模型提取药物和靶点的初始表示,采用轻量级注意力机制和适当的特征交互模块,结合对比学习策略进行训练。实验使用BindingDB、Davis等多个公共数据集,对比AFN、DCAP等经典模型。结果显示,DrugKANs在各数据集上的AUC等指标表现优异,在酶数据集上表现最佳;参数实验表明模型对参数变化相对不敏感;消融实验证明关键组件对性能提升有重要作用。该模型为药物研发提供了有效的预测工具,但KAN技术的计算复杂性等问题有待进一步研究。
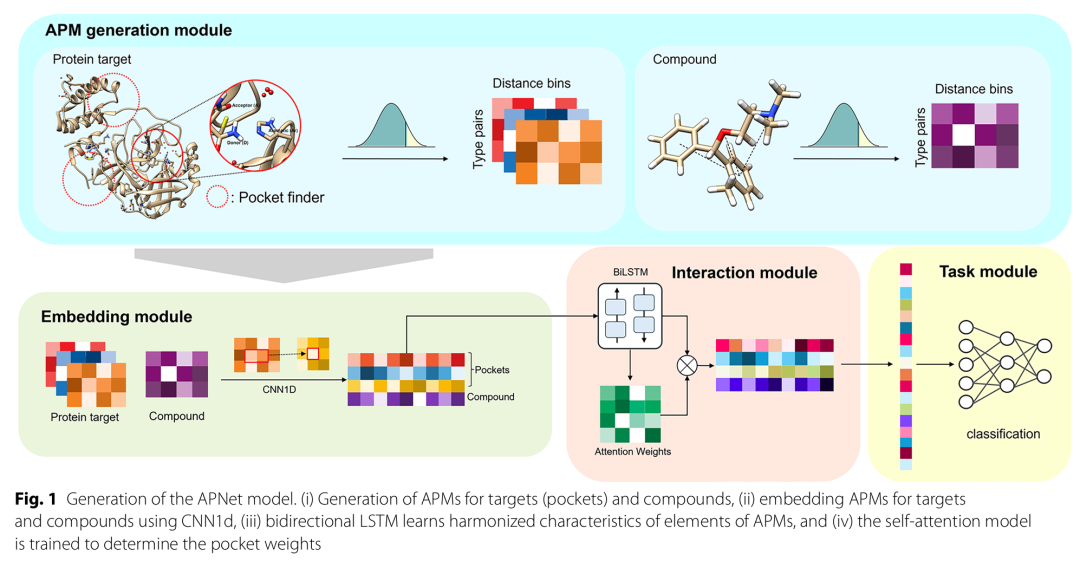
10. Application of 3D atom pair map in an attention model for enhanced drug virtual screening
期刊: Journal of Cheminformatics 链接: https://doi.org/10.1186/s13321-025-01023-2 代码: https://github.com/rimeless/APM
简介: 该研究提出3D原子对图(APM)及基于它的注意力模型APNet,用于药物虚拟筛选,APM能编码分子3D空间信息,优于传统分子表示方法。研究构建APNet模型,包含APM生成、嵌入、交互和任务四个模块,以APM为输入进行化合物-靶点相互作用预测。实验使用PubChem、BindingDB等多个数据集,对比APM与指纹、分子图等其他分子表示方法及相关模型。结果表明,APM在区分活性配体和非活性配体、预测生物测定活性等方面表现出色,基于APM的APNet模型在多个基准测试中性能优于其他模型。该研究为药物虚拟筛选提供了更有效的分子表示和模型,对药物发现具有重要意义。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-05-09,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号