分子生成模型 TABASCO:用极简架构实现物理级精度,速度提升 10 倍!

分子生成模型 TABASCO:用极简架构实现物理级精度,速度提升 10 倍!

MindDance
发布于 2026-01-08 12:54:06
发布于 2026-01-08 12:54:06
在药物研发和材料科学领域,3D分子生成技术一直是AI驱动创新的核心战场。传统模型依赖复杂的对称性约束和图神经网络,却始终难以平衡生成效率与物理真实性。今天,我们要介绍的TABASCO模型彻底打破了这一僵局——它用最简单的Transformer架构,在GEOM-Drugs基准测试中刷新了PoseBusters有效性纪录,同时将推理速度提升了10倍!
颠覆认知:放弃“祖传”设计,反而更高效?
过去的3D分子生成模型普遍遵循三大设计原则:
- • SE(3)等变对称性:确保分子旋转/平移后输出一致
- • 图神经网络(GNN):通过消息传递捕捉原子间相互作用
- • 复杂流匹配目标:依赖耦合最优传输等高级数学框架
但这些“祖传”设计并没有解决核心问题——生成的分子仍频繁出现化学键长度异常、空间位阻冲突等物理不合理性。
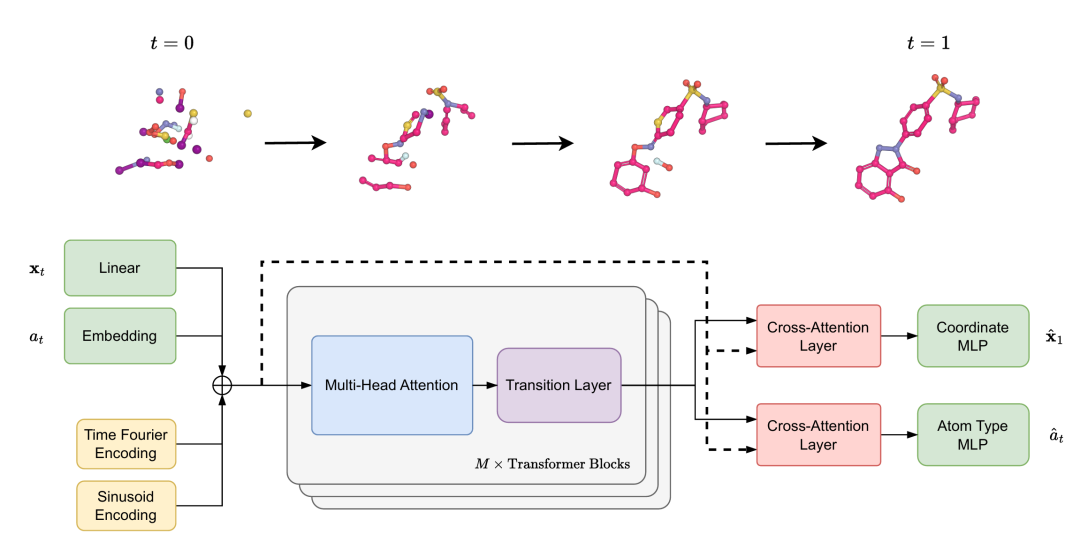
上半部分为 “噪声 - 数据插值过程”,下半部分为 “TABASCO 模型架构”
上半部分为 “噪声 - 数据插值过程”,下半部分为 “TABASCO 模型架构”
上半部分:从噪声到分子的连续蜕变
图中上半部分展示了TABASCO的生成逻辑:分子是从纯噪声(高斯分布)通过连续插值逐步演化而来。这个过程遵循流匹配(Flow-Matching)框架,通过学习时间依赖的速度场,将噪声“运输”到真实分子分布。
具体来说,对于时刻(),分子坐标()由噪声()和目标分子()插值得到:()(其中()为高斯噪声)。模型的任务是预测从()到()的演化速度,最终实现从噪声到完整分子的“平滑过渡”。
下半部分:Transformer如何“看懂”分子
图2下半部分是TABASCO的核心架构,可拆解为三大模块:
- 1. 输入编码层
- • 原子坐标:通过无偏线性层映射到模型隐藏维度
- • 原子类型:通过嵌入层编码(涵盖C、N、O等常见元素及杂原子“*”)
- • 时间信息:采用傅里叶编码捕捉时间依赖特征
- • 位置编码:基于SMILES序列的正弦编码,传递原子的相对顺序信息
这些编码并非简单拼接,而是直接相加形成联合表征——这种设计大幅减少了参数冗余,却保留了关键信息。
- 2. Transformer主体
由16个标准Transformer块堆叠而成(不同规模模型保持一致),每个块包含:
- • 层归一化 + 多头自注意力
- • 层归一化 + 前馈网络(带残差连接)
与传统分子模型不同,这里完全没有使用图注意力或等变层,仅通过自注意力捕捉原子间的长程依赖。
- 3. 输出头
通过两个并行的交叉注意力层分别预测:
- • 原子坐标:经无偏MLP输出3D坐标(优化目标为欧氏条件流匹配)
- • 原子类型:通过离散流匹配框架(DFM)预测元素种类
这种“分离头”设计让模型能针对性优化连续坐标和离散类型,避免任务间的干扰。
TABASCO的叛逆之处在于:它完全抛弃了这些约束,采用非等变Transformer架构,将原子视为序列处理,生成后再用化学工具确定性重建化学键。这种“减法思维”带来了惊人效果:
- • 模型架构简化60%以上
- • 数据吞吐量大幅提升
- • 在GEOM-Drugs数据集上,PoseBusters有效性达到0.92(SOTA)
- • 推理速度比最强基线快10倍
核心创新:四大突破重新定义分子生成
1. 无键设计:让Transformer专注于坐标生成
传统模型将化学键作为独立模态处理,导致计算资源分散。TABASCO反其道而行:
- • 生成时只预测原子坐标和类型,不建模化学键
- • 生成后用RDKit等成熟工具根据坐标自动推断键合关系
- • 实验证明:只要坐标足够精确,化学键信息可完全由物理规则推导
这种设计让模型将90%的计算资源集中在坐标生成上,大幅提升精度。
2. 物理约束微调:最后一公里的质量保障
即使是优秀模型,生成后期也可能出现微小坐标漂移,导致物理规则违反。TABASCO的距离边界引导机制解决了这一问题:
- • 预计算元素对的距离上下限(基于范德华半径和共价键长度)
- • 在采样后期(t=0.99)启动约束优化,修正偏离的原子位置
- • 无需力场计算,仅通过简单梯度下降实现
效果:PoseBusters有效性从0.92提升至0.94,接近训练数据的物理质量(0.94)。
表:物理约束引导 vs 传统力场优化的效果对比
方法 | PoseBusters有效性 | 多样性 | 应变能 | 采样时间 |
|---|---|---|---|---|
无引导 | 0.91 | 0.88 | 14.16 | 10.67s |
距离约束引导 | 0.94 | 0.89 | 19.23 | 75.65s |
UFF力场优化 | 0.94 | 0.88 | 4.74 | 14.21s |
3. 序列编码:SMILES隐藏的空间密码
分子没有天然线性顺序,但TABASCO发现:SMILES字符串的原子排序蕴含空间信息。通过:
- • 基于SMILES序列的正弦位置编码
- • 捕捉序列中相邻原子的空间 proximity
- • 辅助模型在生成早期建立粗结构框架
实验证明,有无位置编码的模型性能差异显著:
- • 带编码:PoseBusters有效性0.91
- • 无编码:有效性暴跌至0.70,出现大量空间碎片化分子
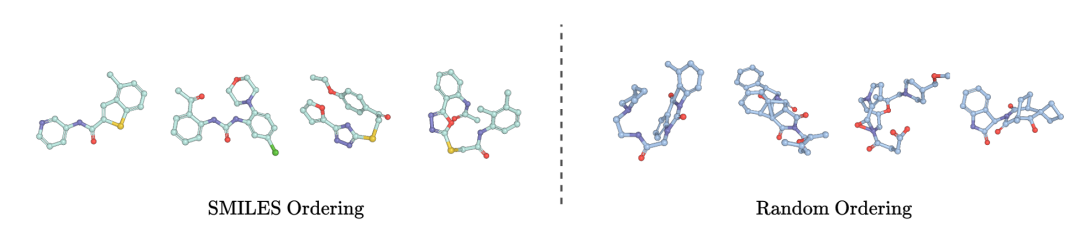
左为带SMILES位置编码的分子结构(完整连贯),右为无编码的碎片化结构,体现序列信息对早期生成的关键作用
左为带SMILES位置编码的分子结构(完整连贯),右为无编码的碎片化结构,体现序列信息对早期生成的关键作用
4. 涌现的对称性:没有硬编码,却自发学会等变
最令人惊讶的是,尽管没有SE(3)等变层,TABASCO却表现出涌现的旋转等变性。通过测量旋转后的预测偏差(ε_equiv)发现:
- • 误差随去噪过程逐渐降低(t→1时趋近于0)
- • 带位置编码的模型误差比无编码模型低一个数量级
这表明:足够的数据和简单架构,能让模型自发学习物理对称性,无需硬编码约束。
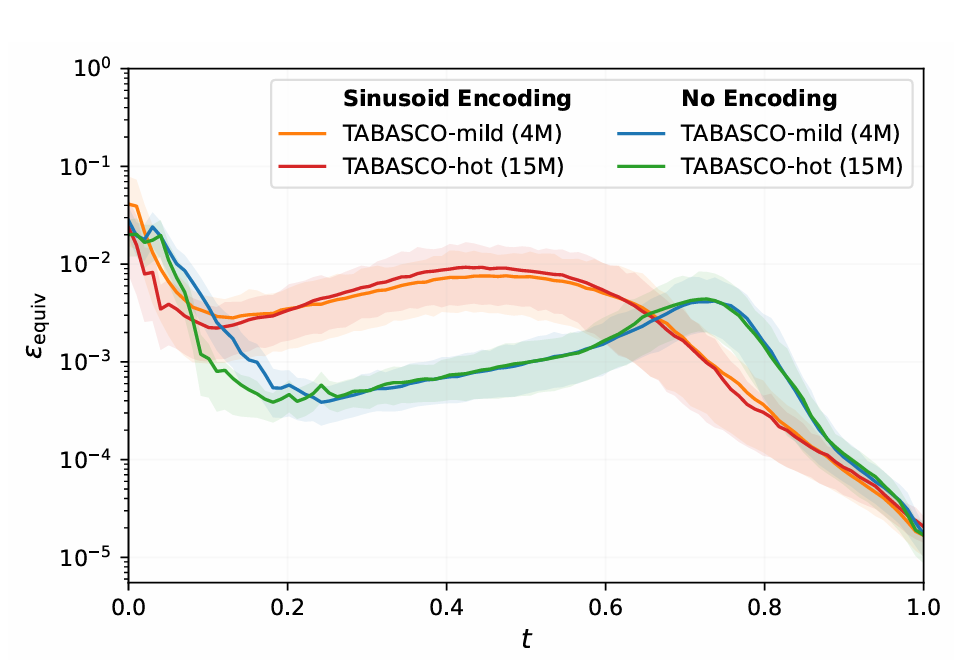
不同t值下的等变误差对比,带位置编码的模型误差始终更低,且随t增大快速下降
不同t值下的等变误差对比,带位置编码的模型误差始终更低,且随t增大快速下降
性能碾压:当简化遇上高效,数据不会说谎
在GEOM-Drugs基准测试中,TABASCO各版本全面领先:
模型 | 参数规模 | PoseBusters有效性 | 多样性 | 采样速度 |
|---|---|---|---|---|
FlowMol | 4.3M | 0.64 | 0.91 | 较慢 |
SemlaFlow | 22M | 0.88 | 0.91 | 中等 |
TABASCO-hot | 15M | 0.91 | 0.88 | 快10倍 |
TABASCO-spicy | 59M | 0.92 | 0.89 | 快10倍 |
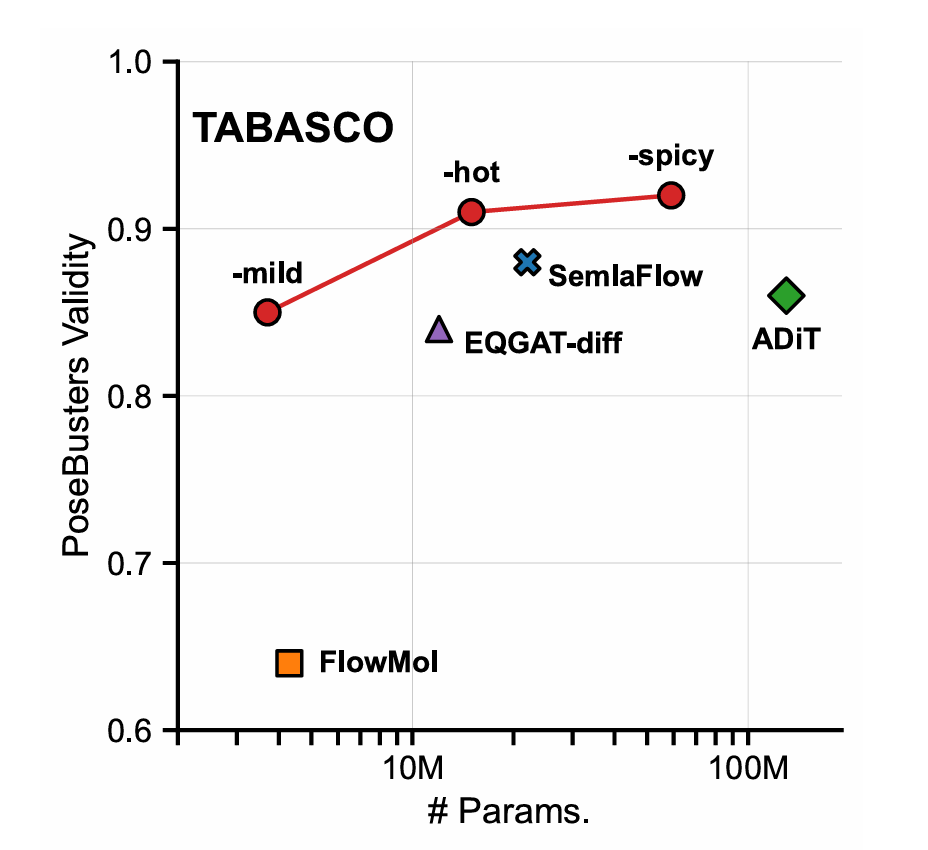
各模型的PoseBusters有效性对比,TABASCO-spicy以0.92显著领先,且参数规模仅59M
各模型的PoseBusters有效性对比,TABASCO-spicy以0.92显著领先,且参数规模仅59M
值得注意的是,15M参数的TABASCO-hot已实现91%的有效性,继续增大参数收益递减,证明极简架构的高效性。
未来展望:从分子生成到药物研发的落地
TABASCO的设计为药物研发提供了新范式:
- • 高吞吐量筛选:10倍速提升让大规模虚拟筛选成为可能
- • 基于药效团的设计:精确的3D结构生成适配靶点结合口袋
- • 可扩展性:模型架构简单,易于集成条件生成和强化学习优化
目前团队已开源代码(github.com/carlosinator/tabasco,不过似乎还未上传),为行业提供了一个即插即用的分子生成工具。
结语:少即是多的AI化学哲学
TABASCO的成功印证了一个道理:在分子生成领域,复杂不等于有效。通过剥离冗余约束,聚焦物理本质,反而能实现精度与效率的双赢。这不仅是技术突破,更重塑了我们对AI建模化学系统的认知——有时候,让模型“轻装上阵”,才能跑得更快更远。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-07-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号