口服小分子为何仍然是最难、也最值得被 AI 改写的药物形态

口服小分子为何仍然是最难、也最值得被 AI 改写的药物形态

MindDance
发布于 2026-05-15 14:14:08
发布于 2026-05-15 14:14:08
本文改写、解读自 Sylvain Gariel 的长文 Patients Want Pills。 原文作者:Sylvain Gariel
过去几年,AI 制药最常见的叙事是:
AlphaFold 改写了蛋白质结构预测,RFdiffusion、ProteinMPNN、抗体设计模型让蛋白质和抗体发现越来越像一条工程化流水线。模型越来越大,结构预测越来越准,生成能力越来越强,很多过去需要几年才能完成的任务,正在被压缩到几周、几天,甚至一次 API 调用。
但药物最后不是给模型吃的。
药物是给患者用的。
所以这篇文章最有意思的地方,是从一个非常朴素的问题开始:
如果疗效和安全性接近,一个糖尿病或肥胖患者,是更愿意每周打一针,还是每天吃一片药?
很多人会选药片。
这并不是说注射剂不重要。过去几年,GLP-1 注射剂已经在糖尿病和肥胖治疗中证明了巨大的临床价值和商业价值。长效注射、抗体、细胞治疗、核酸药物,也都在各自疾病领域解决了小分子很难解决的问题。
但只要有选择,患者对药片的偏好仍然很强。
对多数口服小分子片剂来说,它通常意味着不需要冷链,不需要针头,不需要输注中心,不需要锐器处理,也不需要把日常生活绑定在复杂的给药体系上。更重要的是,如果副作用难以忍受,药片可以停几天;长效注射剂却很难被收回来。
这就是为什么口服小分子在现代药物发现里仍然有一种特殊地位。
它不是最时髦的 modality,却可能是最接近规模化医疗的 modality。真正能触达大规模人群的,不一定是最复杂、最先进、最昂贵的治疗方式,而往往是那些多数情况下可以常温运输、可大规模生产、可由患者自主服用的口服药。
问题是:
既然药片这么重要,为什么我们没有更多真正好的口服药?
这篇文章的核心回答是:
不是因为行业不想做,而是因为口服小分子药物发现,在结构上就比很多人想象得更难。
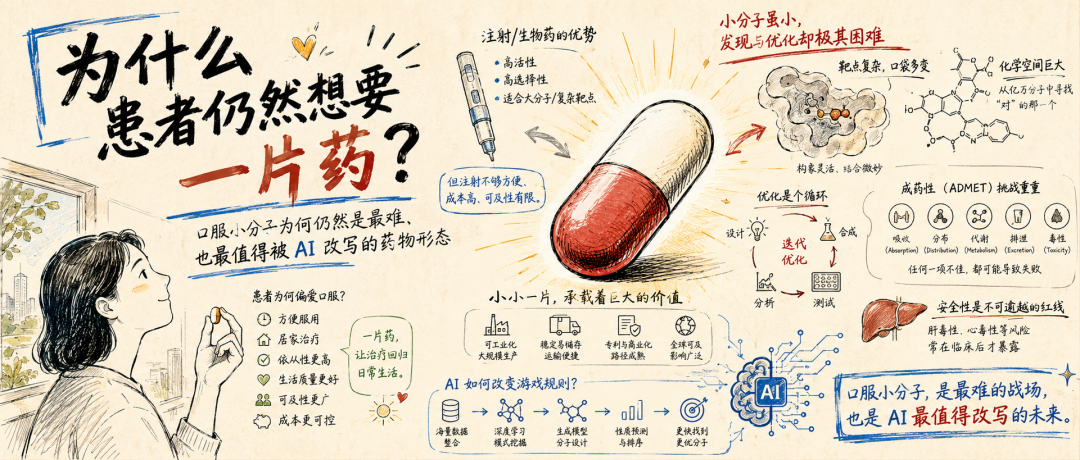
结论先行:
第一,药片不是一种过时的给药方式。 恰恰相反,它可能是最能规模化触达患者的药物形态之一。口服 GLP-1 的监管和商业进展,已经说明患者和市场都在认真回应这种需求。
第二,抗体发现正在变得越来越平台化,但口服小分子没有经历同等程度的成本坍缩。 对很多已经验证过的靶点,抗体发现的前端流程越来越工程化;而口服小分子仍然高度依赖药物化学、ADMET、安全性和合成迭代。
第三,小分子的难点不是只要解决结合就行。 一个能成为药的口服小分子,必须同时通过至少六道关:结合、选择性、通透性、代谢稳定性、安全性、合成可行性。
第四,抗体和小分子的核心差异,可以理解为一种选择性的倒置。 抗体通常更容易获得选择性,但分布能力受限;小分子可以进入细胞、组织甚至中枢,但也因此更容易产生脱靶和毒性问题。
第五,AI 已经在小分子发现中产生真实价值,但它还没有解决最贵、最致命的全部问题。 生成式化学、虚拟筛选、蛋白家族内选择性预测、早期 ADMET 预测和自动化合成闭环都在进步;但靶点选择、人体毒性、罕见安全风险,仍然是 AI 最难直接解决的地方。
第六,真正的验证不在 benchmark,而在临床。 Rentosertib 已经给出了 AI-originated 小分子的早期临床概念验证;zasocitinib 则是 physics-plus-ML 小分子发现走向关键 Phase 3 读数的重要案例。但最终的判断标准只有一个:能不能获批,能不能成为患者真正用得上的药。
一、药片是一种医疗基础设施
我们先从 GLP-1 说起。
Rybelsus 是较早商业化的口服 GLP-1 药物,用于 2 型糖尿病。2025 年,Novo Nordisk 年报显示,Rybelsus 销售额达到 220.93 亿丹麦克朗。2025 年 12 月,Novo Nordisk 又宣布 Wegovy 口服片剂在美国获批,用于长期体重管理和降低特定心血管风险,并预计 2026 年 1 月在美国上市。
2026 年 4 月,FDA 又批准了 Lilly 的 Foundayo,也就是 orforglipron。它是一种每日一次口服的 GLP-1 受体激动剂,也是一个非肽类小分子口服 GLP-1 药物。
这些节点说明了一件事:
即使在长效注射剂已经非常成功的今天,口服药仍然是一个巨大的需求。
原因并不复杂。
药片可以常温运输,可以放进普通药房,可以由患者自己服用,可以覆盖更广泛的人群。它不依赖输注中心,不依赖专业医护人员完成给药,也不需要像许多生物药那样依赖冷链和复杂配送体系。
从医疗可及性的角度看,药片不是先进疗法的低级版本。
它更像是一种基础设施。
真正困难的不是让一个药在少数患者身上产生效果,而是让它以可接受的成本、安全性和便利性,进入足够多患者的日常生活。
这也是为什么患者想要药片这句话并不只是消费偏好,而是一个关于医疗系统可扩展性的判断。
二、那为什么我们没有更多口服药?
如果药片这么重要,为什么很多治疗领域仍然高度依赖注射剂、输注疗法或复杂生物药?
答案不是药企不想做。
答案是:口服小分子太难做。
过去二十年,抗体发现正在变得越来越平台化。展示技术、转基因动物、抗体工程和 AI-native 抗体设计,让一个相对成熟的团队更容易从验证靶点走到可开发候选物。
抗体当然也很难。它有免疫原性、稳定性、组织分布、生产工艺和成本问题。但在很多靶点上,抗体最核心的优势是:它很擅长选择性结合。
这不难理解。抗体有很大的接触界面,可以用一个复杂表面去识别另一个复杂表面。对许多细胞外靶点来说,只要靶点本身足够清楚,找到一个能结合、且相对选择性好的抗体,已经越来越像一个平台化问题。
小分子刚好相反。
小分子的优势在于它能去抗体去不了的地方。
设计得当的小分子有机会穿过细胞膜、进入细胞内,甚至在特定情况下穿过血脑屏障;它也更适合覆盖许多抗体不容易处理的靶点,例如胞内激酶、转录因子、细胞内信号蛋白,以及大量依赖小分子成药的 GPCR 和离子通道靶点。
但代价是选择性。
一个能到处去的小分子,也可能到处乱结合。
人体内有大量潜在可成药蛋白,其中很多蛋白口袋在化学空间里长得并没有那么不同。一个小分子如果可以进入许多组织、许多细胞、许多蛋白口袋,它就必须证明两件事:
我能打中目标。
我不会打中太多不该打的东西。
这就是原文里非常重要的一个概念:选择性的倒置。
抗体用分布能力换选择性。 小分子用选择性换分布能力。
如果最大问题是能不能足够选择性地打中这个靶点,抗体往往更舒服。 如果最大问题是药物能不能到达这个地方,小分子往往更有机会。
真正难的靶点,往往刚好卡在中间:
它们在细胞内,所以抗体碰不到。 它们的口袋又浅、短暂、难以区分,所以小分子也很难做。
你想要小分子的分布能力,又想要抗体的选择性。
这就是口服小分子药物发现的残酷之处。
三、口服小分子不是一道题,而是六道题叠在一起
很多人对 AI 制药的直觉是:
模型生成一个分子,分子打中靶点,然后药就出来了。
真实情况要复杂得多。
一个能成为药的口服小分子,至少要通过六道关。
第一,它要能结合靶点。 也就是 potency 要够,否则剂量不可接受。
第二,它要足够选择性。 不能把一堆相似蛋白都打了。
第三,它要能被吸收。 吃进去之后,要穿过胃肠道屏障,进入血液,再到达目标组织。
第四,它要有合适的代谢稳定性。 不能刚进身体就被肝脏清掉,也不能稳定到在体内长期堆积。
第五,它要安全。 不能有明显 hERG 风险,不能产生危险反应性代谢物,也不能在人体中暴露出难以预测的严重毒性。
第六,它要能合成。 路线要可行,成本要可控,规模要能放大,CMC 要能经受监管审查。
这六件事,每一件都是一个专业领域。
更麻烦的是,它们并不独立。
你为了提高 potency 加了一个基团,溶解度可能崩了。 你为了提高代谢稳定性换了一个结构,可能引入新的毒性风险。 你为了改善溶解度做了极性修饰,通透性可能下降。 你为了提高选择性微调某个原子,合成路线可能变得昂贵又脆弱。
小分子没有这个部分负责结合,那个部分负责开发性的清晰分工。
每一个原子都在同时做多件事。
这就是为什么药物化学很难被简单自动化。它不是在一个目标函数上爬坡,而是在一个高度耦合、非凸、充满隐藏失败模式的空间里寻找少数可行解。
四、最重要的变量,常常是起始 scaffold
在小分子发现里,很多项目并不是死在完全没有 hit。
它们死在有东西,但每个东西都差一点。
差一点活性。 差一点选择性。 差一点溶解度。 差一点通透性。 差一点半衰期。 差一点安全边际。 差一点合成可行性。
而这些差一点,可能会让一个项目从两年拖到六年。
原文里有一个判断非常关键:
在 hit-to-lead 阶段,起始 scaffold 的质量,可能比任何单一变量都重要。
好的 chemical matter,会让后面的优化像一条相对顺滑的路:potency 可以往上推,ADMET 比较干净,选择性有优化空间,合成路线还能承受放大。
差的 chemical matter,则会变成多年折磨:每次修一个问题,又带出另一个问题。活性做上去了,安全性崩了;安全性修好了,选择性没了;选择性改善了,合成路线又不可放大。
这也是为什么 AI 生成分子本身并不等于药物发现完成。
真正的难点不只是生成一个看起来像药的分子,而是更早识别:
这个 scaffold 有没有未来?
它是不是值得投入后面几百个化合物、几十轮合成和数千万美元?
这也是 AI 真正有价值的地方之一。它未必要一开始就给出最终药物,但如果它能更早排除坏 scaffold,更快找到值得优化的化学起点,就已经能改变项目经济学。
五、合成速度决定迭代速度
小分子发现是一门实验科学,不是纯计算游戏。
模型可以在电脑里生成一万个候选分子,但项目真正往前走,仍然要靠设计、合成、测试、反馈,再设计、再合成、再测试。
虚拟筛选可以很快。 真实合成很慢。 真实测试也很慢。
在 hit finding 阶段,大型虚拟库和现货化合物库确实能让团队快速拿到一批候选。但一旦进入 hit-to-lead 和 lead optimization,问题就会重新回到真实化学世界:新的 scaffold、复杂立体化学、定制 bioisostere、路线开发、纯化、稳定性、放大。
这也是为什么自动化合成和闭环实验很重要。
AI 如果只停留在生成端,价值有限。它必须进入一个更完整的闭环:
生成候选。
判断哪些值得合成。
自动或半自动合成。
快速测试。
把实验数据喂回模型。
继续下一轮优化。
药物发现里真正类似 self-play 的地方,不是模型在屏幕上不断生成更多假设,而是模型能决定下一步最值得做哪个实验。
因为提高假设数量并不会自动产生药物。 提高验证质量,才可能产生药物。
六、真正杀死项目的,常常发生在人体里
即使一个小分子通过了早期筛选,进入临床,也不代表它真正安全。
小分子有一个长期难题:DILI,也就是 drug-induced liver injury,药物诱导肝损伤。
一部分 DILI 是剂量相关的,动物实验和早期临床比较容易发现。更难的是特异质性 DILI。它可能和免疫机制、代谢物、个体差异有关,发生率很低,却可能在 Phase 3 或上市后暴露出来。
这类风险对口服小分子尤其残酷。
因为许多小分子都要经过肝脏代谢。只要某个 scaffold 产生了危险的反应性代谢物,或者干扰胆汁酸转运、线粒体功能等关键机制,就可能从有效药物变成临床灾难。
原文用 Takeda 的 fasiglifam 作为例子。
Fasiglifam,也就是 TAK-875,是一个口服 GPR40 激动剂,曾经用于 2 型糖尿病开发。这个药并不是没有疗效。真正让项目停止的,是后期临床中出现的肝脏安全性信号。Takeda 在 2013 年宣布终止 fasiglifam 的开发,理由是综合临床数据后,获益风险不再支持继续推进。
后续公开综述也显示,fasiglifam 组 ALT 升高发生率明显高于安慰剂和活性对照组,并识别出 Hy’s Law 或接近 Hy’s Law 的严重肝损伤案例。
这件事说明了一个残酷现实:
对口服小分子来说,临床不是验证模型的地方。
临床是审判模型的地方。
如果一个模型只能预测早期活性,却不能更早发现后期安全风险,它仍然可能帮你更快地走向失败。
七、AI 到底改变了什么?
那么,AI 能不能解决这些问题?
答案不是简单的能或不能。
更准确地说:
AI 正在改变小分子药物发现的一部分,但还没有改变最难的全部。
目前看,AI 已经在几个环节产生了真实价值。
生成式化学模型可以根据口袋提出更像药的分子。 在结构和活性数据密集的蛋白家族里,选择性预测正在变得更有用。 早期 ADMET 指标,比如溶解度、通透性、微粒体稳定性、CYP 抑制等,正在被模型更好地预测。 虚拟筛选已经不再只是看起来很聪明,而是开始能帮助团队更有效地 triage。 自动化合成平台也在把设计—合成—测试的周期从几个月压缩到几周。
但这些进步并不等于AI 一键生成新药。
更像是:
AI 开始进入药物发现的真实工作流,帮助团队更快迭代、更少浪费合成轮次、更早排除明显不值得推进的分子。
这很重要,但它不是终点。
因为真正杀死许多项目的两个问题,仍然是:
靶点选错。
人体毒性不可接受。
前者决定这个药有没有生物学意义。 后者决定这个药有没有临床未来。
这两件事,今天仍然很难被 AI 直接解决。
八、临床信号开始出现了
AI 制药真正重要的不是论文,也不是 benchmark,而是临床数据。
这里有三个值得关注的案例。
1. Rentosertib:AI-originated 小分子的早期临床概念验证
Rentosertib 是 Insilico Medicine 开发的 TNIK 抑制剂,用于特发性肺纤维化。它的重要性在于,TNIK 这一靶点和 rentosertib 这一分子,都是由 Insilico 的 AI 平台推动发现和设计的。
2025 年,Rentosertib 的 Phase 2a 结果发表在 Nature Medicine。该研究是一项多中心、双盲、随机、安慰剂对照试验。公开数据中,60 mg QD 剂量组在 12 周时显示 FVC 平均增加 98.4 mL,而安慰剂组平均下降 20.3 mL。
这不是最终胜利。
Phase 2a 的样本量和时间都有限,后续仍然需要更大规模、更长周期的验证。但它至少说明一件事:
AI-originated 的靶点和小分子,已经可以走到临床概念验证阶段。
这和停留在 benchmark 上完全不是一回事。
2. Garutadustat:平台可重复性的观察点
Garutadustat 是 Insilico 另一个 AI 驱动的小分子项目,是一个用于炎症性肠病的肠道限制性 PHD 抑制剂。根据公司公告,该项目已经进入 Phase IIa 给药阶段。
它目前还不是疗效验证案例,更不能被写成成功药物。
它真正值得关注的地方是:
如果同一平台不只偶然推进一个项目,而是能反复把候选物推到临床阶段,行业才会开始相信它的可重复性。
AI 制药最需要证明的不是我做成了一个漂亮案例。
而是:
我能不能稳定地产出临床级候选物?
3. Zasocitinib:最接近审批验证的计算驱动小分子案例之一
Zasocitinib,也就是 TAK-279,是 Takeda 从 Nimbus 获得的选择性 TYK2 抑制剂,用于中重度斑块型银屑病。这个项目与 Schrödinger 的 physics-plus-ML 平台密切相关,核心不是端到端生成式 AI,而是物理建模、自由能扰动计算和机器学习辅助的小分子优化。
2025 年 12 月,Takeda 宣布 zasocitinib 的两项关键 Phase 3 研究达到主要和排序次要终点。公开结果显示,超过一半接受 zasocitinib 治疗的患者在第 16 周达到 PASI 90,大约 30% 达到 PASI 100。
这个案例的重要性在于:
它不是一个概念验证阶段的故事,而是一个已经进入关键 Phase 3 数据读数、准备监管申报的计算驱动小分子案例。
但这里也要说清楚:
Zasocitinib 不是端到端生成式 AI 发现药物。
它更像是 physics-plus-ML 在小分子药物化学中产生真实临床价值的案例。
如果它最终获批,它验证的不是AI 可以替代药化,而是:
在某些足够明确、数据足够密集、物理模型足够有用的问题上,AI 和物理建模可以真正提高小分子发现效率。
九、ADMET 数据正在成为新的战场
如果说小分子药物发现最缺什么,很多人会说是更好的模型。
但更深层的问题可能是:
更好的 ADMET 和安全性数据。
药企最宝贵的数据,往往不是公开结构,也不是论文里的活性数字,而是内部多年积累下来的失败数据、毒性数据、代谢数据、开发性数据。
这些数据过去高度封闭。
因为它们直接关系到研发竞争力。
但现在,行业开始出现一些新的数据合作方式。
Lilly 在 2025 年推出 TuneLab,让外部 biotech 可以使用基于 Lilly 大规模内部研究投入训练的 AI 模型,同时通过联邦学习等方式保护数据边界。
Inductive Bio 等公司则在 ADMET 预测竞赛中取得进展,说明早期开发性预测正在从经验辅助走向更系统的模型能力。
Apheris 也在推动面向 ADMET 的联邦数据网络,让多家药企能够在不直接暴露原始数据的情况下共同训练模型。
这些事情听起来没有AI 生成新药那么性感。
但它们可能更接近小分子的真实瓶颈。
因为对口服小分子来说,真正稀缺的不是又一个候选分子,而是更早知道:
哪个分子会死。
为什么会死。
死在药代、毒性、代谢、相互作用,还是合成放大。
如果 AI 能把这些问题提前暴露出来,它的价值会非常大。
十、监管也在变,但不要误读成动物实验已经过时
监管环境也在变化。
2025 年,FDA 发布了减少临床前安全性研究中动物测试的路线图,提出引入新方法,包括 AI 计算模型、类器官、organ-on-chip 等方式。不过这一路线图的初始重点是单克隆抗体,之后才会扩展到其他生物分子和新化学实体。
2026 年 1 月,FDA 和 EMA 又联合发布了 AI 在药物开发中良好实践的指导原则,强调以人为本、基于风险、数据治理、模型生命周期管理、透明性等原则。
这些变化很重要。
但它们不是制度上的一夜转向。
尤其对新化学实体,也就是大量口服小分子来说,标准毒理包、动物实验和传统安全性验证仍然不会很快消失。
原因也很简单:
科学还没有完全准备好。
如果我们还不能可靠预测人体毒性,监管就不可能只因为模型看起来先进,就放弃已有安全框架。
路线图是方向,不是终点。
十一、AI 还没有解决什么?
这篇文章最值得借鉴的地方,是它没有把 AI 制药讲成一个线性乐观故事。
它承认进展,也保留怀疑。
AI 已经能帮我们更快生成分子,更好做早期筛选,更有效地做 ADMET triage。
但它还没有真正解决长尾问题。
比如特异质性 DILI。 真实阳性样本很少,机制高度异质,传统数据集很难支撑模型学习。一个模型可以在总体准确率上看起来很好,但如果它永远预测安全,它仍然可能错过最重要的少数危险案例。
再比如长期安全性、特殊人群、药物相互作用、罕见代谢路径。
还有 beyond rule-of-five 的问题,比如 macrocycles、PROTACs、molecular glues。它们往往处在口服生物利用度最难处理的区域。
这些问题不是靠多生成一些分子就能解决的。
因为药物发现的瓶颈不只是候选分子不够多。
很多时候,瓶颈是:
我们不知道如何更早判断哪个候选物会在人体里失败。
十二、什么时候我们才知道 AI 真的改变了口服小分子?
原文最后提出了几个判断标准,我觉得非常重要。
第一,要看到多个独立团队,在 hard target 上做出临床阶段口服小分子,而且在 compound count、时间和成本上都有明显改善,同时 PK 和 ADMET 干净。
一次成功可能是幸运。 多次成功才是平台。
第二,要看到真正可前瞻验证的人体毒性预测工具。
不是在历史数据上讲故事,而是能提前预测哪些分子后来会在人体中出现肝毒性或其他严重安全信号。
第三,要看到一个真正 end-to-end AI-discovered 药物进入 Phase 3,并最终获批。
这才是 AI 制药从工具进步到商业验证的分水岭。
换句话说:
AIDD 的终点不是好期刊论文。 不是漂亮 benchmark。 不是融资 PPT。 不是我们生成了十万个分子。
终点是一个患者可以拿到的药。
结语:患者想要药片,行业想要答案
这篇文章的标题叫 Patients Want Pills。
表面看,它讲的是口服小分子的技术难度。
但更深一层,它是在提醒我们:
药物发现不能只从技术供给侧出发,还要从患者需求侧重新看一遍。
患者想要的不是一个 modality。 患者想要的是有效、安全、可负担、可获得、可长期使用的治疗方式。
药片之所以重要,是因为它可能同时满足这些条件。
但也正因为如此,口服小分子才如此困难。
它要进入身体最复杂的分布系统,避开无数相似口袋,经受肝脏代谢和安全性审判,还要能以工业规模制造出来。
这不是一个单纯的生成问题。
这是一场关于结合、选择性、通透性、代谢、安全性和合成的联合优化。
AI 正在进入这场优化。
但它真正改变小分子的方式,可能不是一键生成神药,而是更实际、更缓慢、也更重要的几件事:
更早排除坏分子。 更快找到好 scaffold。 更准确预测 ADMET。 更好结合物理模拟和实验闭环。 更少浪费合成轮次。 更快把真正有希望的分子推进临床。
也许这就是 AI 制药最不浪漫、但最真实的前景:
它不是替代药物发现里的所有不确定性。
它是让我们在不确定性中,少走一些错路。
患者最终并不关心一个药是不是 AI 发现的。
患者关心的是:
它能不能吃。 它有没有效。 它安不安全。 它买不买得起。 它能不能真的来到自己身边。
所以,药物发现里的下一个大问题,不只是 AI 能不能生成更多分子。
而是 AI 能不能帮助我们,把更多真正能用的药片,带到患者手里。
参考文献与资料来源
- Sylvain Gariel,Patients Want Pills,X 长文。本文主体逻辑主要基于该文改写与解读。https://x.com/SylvainGariel/status/2054318185982406698
- Novo Nordisk,Annual Report 2025。其中披露 2025 年 Rybelsus 销售额为 DKK 22,093 million。
- Novo Nordisk,Wegovy pill approved in the US as first oral GLP-1 for weight management,2025-12-22。
- U.S. FDA,FDA Approves First New Molecular Entity Under National Priority Voucher Program,2026-04-01。该公告披露 Foundayo/orforglipron 的批准和适应症信息。
- Eli Lilly,FDA approves Lilly's Foundayo™ (orforglipron) ,2026-04-01。该公告披露 Foundayo 的口服小分子 GLP-1 属性、ATTAIN-1 体重下降数据和上市安排。
- Xu Z. et al.,A generative AI-discovered TNIK inhibitor for idiopathic pulmonary fibrosis: a randomized phase 2a trial,Nature Medicine,2025。
- Insilico Medicine,Nature Medicine Publication of Phase IIa Results Evaluating Rentosertib,2025-06-03。该公告披露 60 mg QD 组 FVC +98.4 mL、安慰剂组 -20.3 mL 等数据。
- Insilico Medicine,Garutadustat Phase IIa Trial First Patient First Dose,2026-01。该公告披露 Garutadustat/ISM5411 进入炎症性肠病 Phase IIa 试验。
- Takeda,Zasocitinib Landmark Phase 3 Plaque Psoriasis Data,2025-12-18。该公告披露 zasocitinib 两项关键 Phase 3 研究达到主要和排序次要终点。
- Takeda,Zasocitinib Delivered Rapid and Durable Skin Clearance in Phase 3 Studies,2026。该公告披露第 16 周 sPGA、PASI 75 等进一步数据。
- Takeda,Takeda Completes Acquisition of Nimbus Therapeutics’ TYK2 Program Subsidiary,2023-02-08。该公告披露约 40 亿美元 upfront 和最高 20 亿美元销售里程碑安排。
- Schrödinger,Design of a highly selective, allosteric, picomolar TYK2 inhibitor using novel FEP strategies。该资料介绍 Nimbus/Schrödinger 在 TYK2 项目中使用 FEP+ 进行选择性和 ADME 相关优化。
- Takeda,Termination of Fasiglifam (TAK-875) Development,2013-12-26。
- Menon V. et al.,Fasiglifam-Induced Liver Injury in Patients With Type 2 Diabetes,Diabetes Care,2018。该研究分析 fasiglifam 临床项目中的肝损伤信号。
- Lilly,TuneLab platform,2025-09。该公告披露 Lilly 向 biotech 开放基于其内部研究投入训练的 AI-enabled drug discovery models。
- Inductive Bio,OpenADMET-ExpansionRx blind challenge win,2026-02-03。
- Apheris,ADMET Network launch,2026-02-25。该公告披露其面向 ADMET 预测的联邦数据网络和 founding members。
- U.S. FDA,Roadmap to Reducing Animal Testing in Preclinical Safety Studies,2025。
- U.S. FDA / EMA,Guiding Principles of Good AI Practice in Drug Development,2026-01-14。
- Marcinak J.F. et al.,Liver Safety of Fasiglifam (TAK-875) in Patients with Type 2 Diabetes: Review of the Global Clinical Trial Experience,Drug Safety,2018。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-14,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号