差异分析的火山图为什么不喷发呢


最近在咱们的微信交流群看到了小伙伴反馈一个2024的一个单细胞数据挖掘文章:《Integration of single‐cell sequencing and bulk RNA‐seq to identify and develop a prognostic signature related to colorectal cancer stem cells》,链接是:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11133358/ ,研究者们重新分析了2011的一个表达量芯片数据集(GSE33113),然后给出来了主要的差异分析结果,火山图就很奇怪:
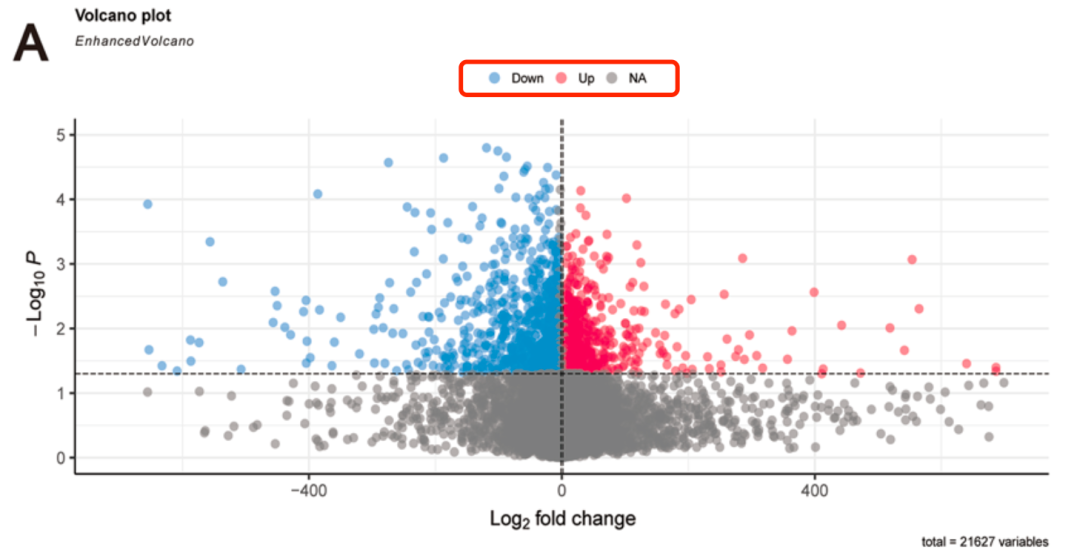
火山图就很奇怪
数据集是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33113
在官方网页可以看到数据集的样品分组不平衡,90 AJCC stage II CRC patients, and matching normal colon tissue from 6 of these patients, 是最常规的表达量芯片平台:Affymetrix Human Genome U133 Plus 2.0 Array
library(AnnoProbe)
library(GEOquery)
gse_number <- 'GSE33113'
if(T){gset <- geoChina(gse_number)}
if(F){ gset = getGEO("GSE33113", destdir = '.', getGPL = F) }
a=gset[[1]]
dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
dim(dat)#看一下dat这个矩阵的维度
dat[1:4,1:4] #查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
table(is.na(dat))
pd = pData(a)
可以很明显的看到作者给出来的表达量矩阵有有瑕疵的;
> table(is.na(dat))
FALSE TRUE
5123412 125388
> dim(dat)
[1] 54675 96
> dat=na.omit(dat)
> dim(dat)
[1] 33777 96
因为这个芯片平台是 54675 个探针,但是只有33777个探针是完整值。
其实上面的简单粗暴的去除有NA值的探针不够细致,更加好的方法是下载这个数据集的cel文件自己走一遍流程。但是上面的33777个探针是完整值仍然是可以对应1.6万个基因,其实在后续分析里面也是绰绰有余了,完全是可以拿到比较符合预期的差异分析结果;
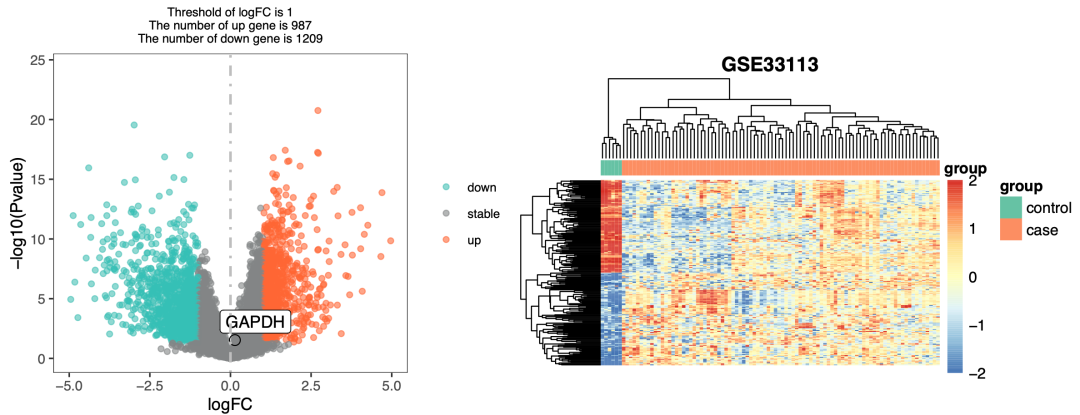
符合预期的差异分析结果
后续的基因上下调差异基因的生物学功能富集,也能看到上调的主要是肿瘤恶性细胞增殖相关的通路,非常符合预期啦。
这个2011的一个表达量芯片数据集(GSE33113)对应的两个文章的关注点都不是差异分析:
- Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011 Nov 4;9(5):476-85. PMID: 22056143
- Mutations in the Ras-Raf Axis underlie the prognostic value of CD133 in colorectal cancer. Clin Cancer Res 2012 Jun 1;18(11):3132-41. PMID: 22496204
因为前面提到了数据集的样品分组不平衡,90 AJCC stage II CRC patients, and matching normal colon tissue from 6 of these patients, 既然是主要是病人的表达量数据,就可以做病人的表达量分子分型,以及预后好坏分析,生存分析等等。
学徒作业
既然是常规的表达量芯片平台:Affymetrix Human Genome U133 Plus 2.0 Array,就可以从GEO界面的cel文件开始自己拿到表达量矩阵文件,然后走差异分析流程拿到上下调基因。
然后上面的代码是直接使用作者的表达量矩阵,虽然里面很多NA值,但是简单粗暴的过滤了NA值之后也正常的走差异分析流程拿到上下调基因。
需要大家比较两次差异分析的结果哦!可以参考前面的系列教程:
写在文末
我在《生信技能树》,《生信菜鸟团》,《单细胞天地》的大量推文教程里面共享的代码都是复制粘贴即可使用的, 有任何疑问欢迎留言讨论,也可以发邮件给我,详细描述你遇到的困难的前因后果给我,我的邮箱地址是 jmzeng1314@163.com
如果你确实觉得我的教程对你的科研课题有帮助,让你茅塞顿开,或者说你的课题大量使用我的技能,烦请日后在发表自己的成果的时候,加上一个简短的致谢,如下所示:
We thank Dr.Jianming Zeng(University of Macau), and all the members of his bioinformatics team, biotrainee, for generously sharing their experience and codes.
十年后我环游世界各地的高校以及科研院所(当然包括中国大陆)的时候,如果有这样的情谊,我会优先见你。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-06-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号