蛋白质语言模型-Big Isn't Always Better

蛋白质语言模型-Big Isn't Always Better

Tom2Code
发布于 2026-04-17 17:29:28
发布于 2026-04-17 17:29:28
"Protein Language Model Choice: Big Isn't Always Better"
先来放上论文地址:
https://doi.org/10.1101/2025.10.30.685515
代码地址:https://github.com/tsenoner/plm_choice
是一篇预印论文
题目是:《which plm to choose?》

作者是来自德国慕尼黑工业大学的Tobias Senoner
plm=protein language model
接下来,我们就通过这篇论文来看看到底应该在计算任务中选择蛋白质语言模型呢?
一.概述
该研究系统地基准测试了十四种蛋白质语言模型(pLMs),旨在评估它们学习到的嵌入如何有效地表征蛋白质的序列、结构和功能相似性。研究发现了一个“规模-性能悖论”,即中等规模的基础模型在直接反映原始嵌入距离中的内在生物信号方面,与体积大得多的模型效果相当。相对而言,大型pLMs展现出卓越的信息提取能力,它们储存了更复杂的可提取信息,但这只有通过额外的监督训练(微调)才能充分利用。作者还指出,针对特定任务的微调会从根本上扭曲嵌入空间的几何结构,从而产生牺牲通用性的专业化表征。因此,该研究建议采用任务驱动的模型选择策略:高效的中等规模模型适用于快速的通用洞察,而最大型的模型则仅在需要微调以达到最高性能时才值得采用。
二.文章回答了怎样的问题?

上图两个问题截取自原文
三.研究材料
3.1数据集
数据集的收集与拆分
研究中准备了两个数据集:主要的 SwissProt-pre2024 数据集和用于评估模型泛化能力的 New2024 数据集。
- SwissProt-pre2024 数据集: 该数据集是用于模型训练、验证和测试的主要数据集,源自 Swiss-Prot 数据库 2024_01 版本。
- 非冗余拆分: 为了创建非冗余的数据拆分,研究人员首先使用 MMseqs2 的聚类模式对所有蛋白质进行了分区。聚类的标准设定为:最小成对序列同一性(PIDE)为 30%(
--min-seq-id 0.3)和较短序列的 80% 覆盖率(-c 0.8,--cov-mode 1)。 - 集合比例: 随后,这些聚类按照聚类大小进行分层拆分,比例为:训练集占 70%,验证集占 15%,测试集占 15%。
- 最终数量: 通过此过程,获得了包含 375,209 个蛋白质的训练集、86,546 个蛋白质的验证集以及 80,514 个蛋白质的测试集(这些都是序列非冗余的蛋白质)。
序列对的组成
蛋白质对的构建是为了建立评估模型时的基础真实值(ground truth)。
- 全对比全搜索: 研究人员使用 MMseqs2 进行了一次全对比全搜索(all-against-all search),以计算数据集中蛋白质对的三种相似性指标:序列同一性、结构相似性和功能相似性。
- 相似性指标:
- 序列相似性以 PIDE(成对序列同一性百分比)报告。
- 结构相似性以 TM-score 报告(使用 Foldseek 计算的 alntmscore)。
- 功能相似性通过 HFSP 分数(同源性衍生蛋白质功能相似性)评估。
- 筛选蛋白质对: 为了确保基础真实数据的高质量,所得的蛋白质对根据已建立的可靠性阈值进行了严格筛选:
- PIDE 筛选: 仅保留 PIDE 大于 0.3 且两个蛋白质的对齐覆盖率都 的蛋白质对。
- TM-score 筛选: 仅保留 TM-score 且两个结构的对齐覆盖率都 的蛋白质对(同时排除了平均预测可靠性 pLDDT 分数 的蛋白质结构)。
- HFSP 筛选: 仅保留 HFSP 分数 大于 0 的蛋白质对。
经过筛选后,最终的测试集(用于所有固有或可提取信息评估)包含 8,731,253 个具有相似序列和功能关系的蛋白质对,以及 15,481,580 个具有相似结构关系的蛋白质对。需要注意的是,这一筛选步骤排除了绝大多数高度不相似的蛋白质对,因此分析主要评估了嵌入(embeddings)定量捕获可测量相似性的能力,而不是二元区分相似与不相似蛋白质的能力。
3.2计算指标
序列相似性(Sequence Similarity): 使用 PIDE (Percent Pairwise Sequence Identity,成对序列同一性百分比) 。
结构相似性(Structural Similarity): 使用 TM-score(通过 Foldseek 计算 alntmscore 获得)。
功能相似性(Functional Similarity): 使用 HFSP(Homology-derived Functional Similarity of Proteins,同源性衍生蛋白质功能相似性)分数 。
3.3实验方法
研究人员建立了一个全面的框架来量化蛋白质语言模型(pLMs)嵌入中编码的生物学信息,主要分为对固有信息和可提取信息的评估 。
以下是实验方法和评估框架的详细介绍:
3.3.1. 被分析的蛋白质语言模型 (pLMs)
研究系统地比较了 14 种常用 pLMs 生成的表示 。这些模型涵盖了不同的架构、训练方法和参数规模 。
- 基础模型 (Foundation models):
- 规模从 800 万 (8M) 到 30 亿 (3B) 自由参数不等。
- 包括 ProtT5 (1.5B 参数) 、ESM-1b (650M 参数) 、ESM-2 的多个变体 (8M, 35M, 150M, 650M, 3B 参数) 、Ankh (450M 和 1.15B 参数) 、ESM-C (300M 和 600M 参数) 和 ESM-3 (1.4B 参数) 。
- 任务特定模型 (Task-specific models):
- 包括 CLEAN (650M 参数),该模型通过对比学习训练以进行酶分类 ;以及 ProtTucker (1.5M 参数),该模型经过优化以进行 CATH 结构域预测 。
嵌入生成: 对于每个蛋白质序列,研究人员通过平均每个 pLM 最终隐藏层的残基表示,生成固定长度的蛋白质级别嵌入,以便进行成对比较分析 。
3.3.2. 评估设置和方法
评估在两种嵌入设置下进行,并采用了两种核心评估方法来区分不同类型的生物信号 :
A. 评估设置 (Embedding Settings)
- 原生嵌入维度 (Native Embedding Dimensions): 使用 pLM 输出的原始维度进行评估 。
- 标准化 128 维表示 (Standardized 128-dimensional representations): 通过 主成分分析 (PCA) 将所有嵌入降维到 128 维后进行评估 。
B. 评估方法 (Assessment Approaches)
1. 固有信息 (Inherent Information)——欧几里得距离固有信息是指可以直接通过对原始嵌入向量进行简单算术计算(例如建立蛋白质距离/相似性)获得的信号。
- 方法: 作为不可训练的基线 ,计算嵌入对之间的欧几里得距离 (Euclidean distances) 。
- 目的: 评估 pLM 嵌入空间本身(原始状态,不进行额外训练)捕捉生物学关系的能力。
2. 可提取信息 (Extractable Information)——前馈网络 (FFN) 监督学习可提取信息是指需要通过后续基于嵌入的监督机器学习(例如预测特定的蛋白质特征)才能揭示的信号。
- 方法: 训练一个前馈网络 (FFNs),使用嵌入对作为输入,以预测生物学属性 。
C. 监督学习 (FFN) 架构和目标
- FFN 架构: 采用对称架构 。每个 维嵌入( 取决于 pLM 且维度不同,例如 128, 1024, 2560)首先通过一个隐藏层 () ,然后将两个嵌入的 64 维输出连接(合并)起来,再通过连续的层 (128 64 32 1) 来生成最终预测 。
- 预测目标 (Targets): FFN 模型被独立训练,以预测蛋白质对的三个相似性指标:
- 序列同一性 (PIDE) 。
- 结构相似性 (TM-score) 。
- 功能相似性 (HFSP score) 。
- 训练细节: 所有模型均使用 PyTorch Lightning 实现,采用一致的超参数,包括学习率 0.001,批量大小 1024,最多 100 个 epoch,以及 5 个 epoch 的早停耐心 。
- 随机对照: 使用从标准正态分布生成的 1024 维随机对照嵌入来建立性能基线 。
3.3.3. 性能和距离分析
- 性能评估指标: 使用 Pearson 相关系数来评估模型预测值与真实值之间的性能(相关性) 。
- 距离分布比较: 为了进行跨模型比较,所有成对嵌入距离都进行了 min-max 归一化,缩放到 范围 。
- Wasserstein 距离 (W): 计算归一化后的成对距离分布之间的 Wasserstein 距离,以量化将一个概率分布转换为另一个所需的最小“成本”,从而比较不同 pLMs 嵌入空间的几何形状 。
四.实验结果
作者在14个蛋白质语言模型中进行了实验,包括不同参数量的:esm1,esm2,esm3,emc,ankh,ProtT5。
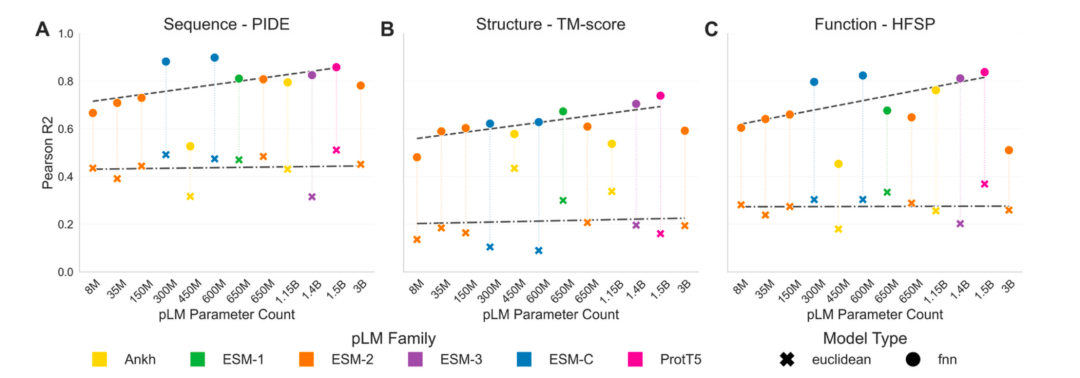
三个图首先分别对应了序列,结构,功能的预测。
每个图又分别对应着两个方法:
首先是 x组成的点,代表了直接计算两个蛋白质向量之间的欧几里得距离与原始数据之间的皮尔逊相关系数。
其次是圆点 ,代表由模型的嵌入层送到一个FFN简单神经网络中,采用固定的网络模型参数和训练参数得到的预测值和真实数据之间的皮尔逊相关系数。
第二个图是验证微调蛋白质语言模型会使得蛋白质语言空间发生形变:
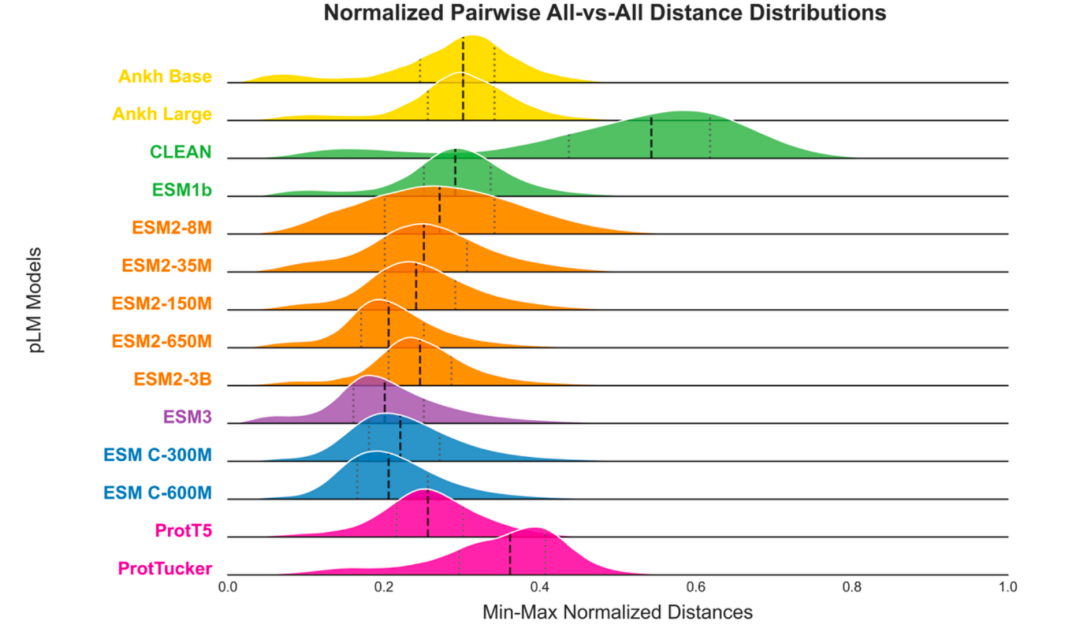
所以有三个关键结论,可以结合上面的图一起看:
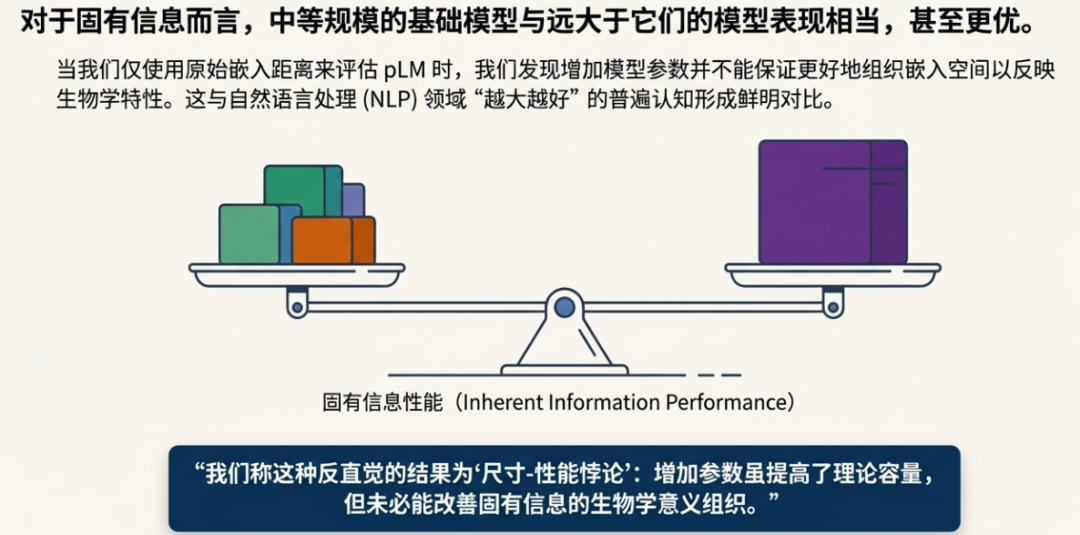
- 规模-性能悖论: 对于仅依赖原始嵌入距离(固有信息)的零样本(zero-shot)应用,中等规模的基础pLM即可达到性能的“甜点”,因为增加数十亿的额外参数并不会提高其内在的生物学意义的组织能力,且消耗更多能源。
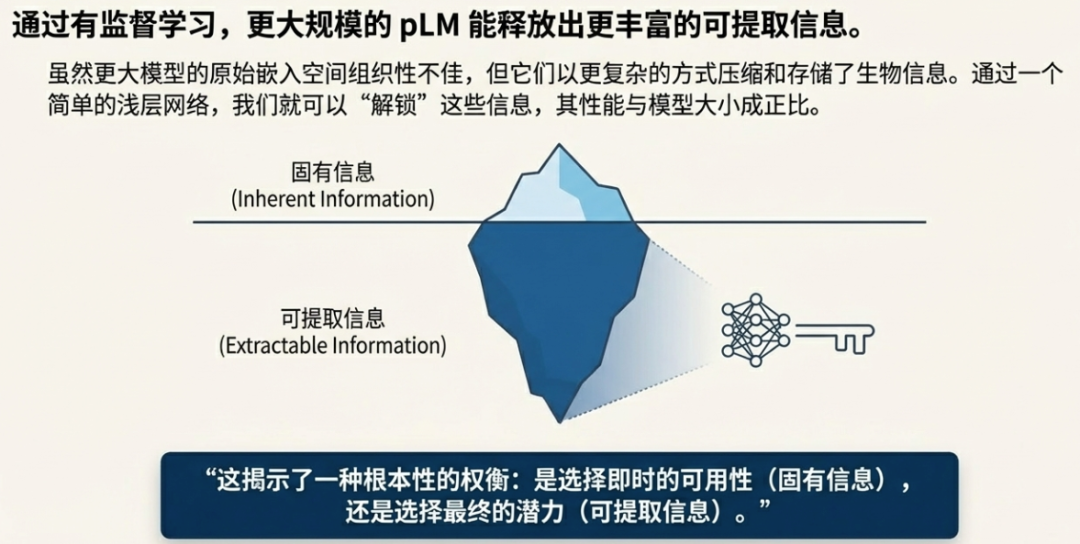
- 可提取信息容量红利: 尽管大型pLM的固有信息组织能力不一定更强,但它们在嵌入中编码了更丰富的蛋白质表征,通过后续的监督学习训练(可提取信息)可以显著提升性能,因此在需要精细调整以实现特定任务时,大模型具有优势。
- 任务特异性训练扭曲几何结构: 针对特定任务(如酶分类或结构域分类)的对比式微调会从根本上重塑嵌入空间的全局几何结构,虽然能提高目标任务的表现,但会牺牲模型的通用性和跨任务泛化能力。
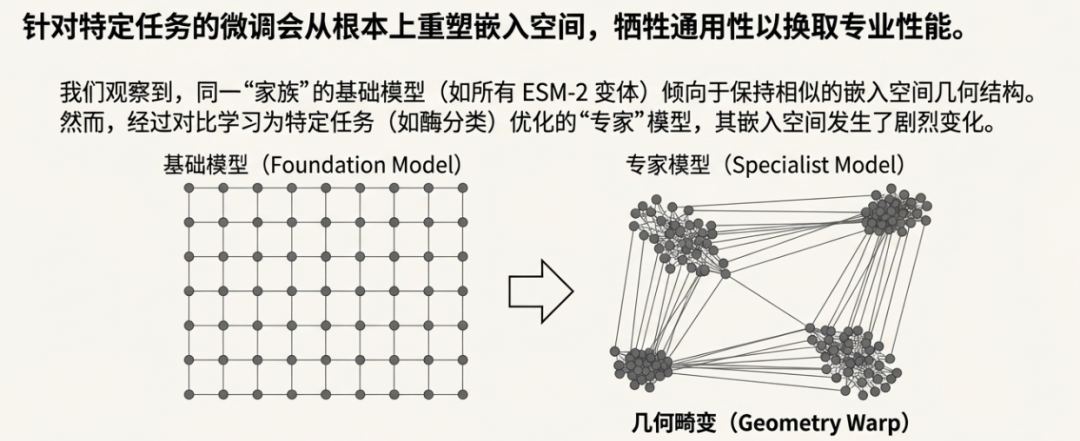
简而言之,应采取任务驱动的模型选择策略:中等规模模型适用于快速、低成本的通用洞察;大型模型适用于计划进行微调以追求最大性能上限的场景;而任务专用模型仅应用于与其训练目标精确匹配的生物学问题。
所以这篇文章最终告诉我们的是:
选择pLM是一场在“即时可用性”与“未来潜力”之间的权衡,蛋白质大语言模型层出不穷,如何选择适合自己的特异性的研究和任务的模型,我相信这是一场关于研究蛋白质语言模型的长途旅行,slowly drive,enjoy the view~
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-12-01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号