三角乘法就是你所需要的一切:Pairmixer 如何重塑生物分子结构预测的计算范式

三角乘法就是你所需要的一切:Pairmixer 如何重塑生物分子结构预测的计算范式

DrugIntel
发布于 2026-05-14 18:00:14
发布于 2026-05-14 18:00:14

论文标题:Triangle Multiplication Is All You Need for Biomolecular Structure Representations 发表会议:ICLR 2026 机构:Genesis Research & UT Austin 代码:https://github.com/genesistherapeutics/pairmixer
一、引言:规模化应用下的计算瓶颈
AlphaFold 系列模型的出现是结构生物学的里程碑事件。然而,随着应用场景从单蛋白质折叠拓展到更复杂的下游任务——高通量虚拟筛选(virtual screening)、蛋白质组规模折叠(proteome-wide folding)、从头蛋白质结合剂设计(de novo binder design)——每一项任务都意味着数以百万计乃至数十亿次的推理调用,计算代价成为不可回避的瓶颈。
以当前 SOTA 模型 Boltz-1(AlphaFold3 的开源复现)为例,在 A100 GPU 上处理一条 2048 token 的序列,单次推理耗时超过 15 分钟。这一数字在批量筛选场景下是灾难性的。
瓶颈的根源在于 AlphaFold3 所引入的 Pairformer 骨干网络(backbone):
- • 配对表示(pair representation) 本身即是 内存复杂度;
- • 建立在其上的 三角注意力(Triangle Attention)需要对配对矩阵的每一行执行独立的注意力计算,共计 次,总计算量达 FLOPs;
- • 即便是相对高效的三角乘法(Triangle Multiplication),也有 的三次方计算量。
面对这一困境,本文提出的核心问题是:三角注意力和序列更新模块,真的是必要的吗?
二、背景知识:Pairformer 架构解析
要理解 Pairmixer 的创新,首先需要理解它所替代的对象。
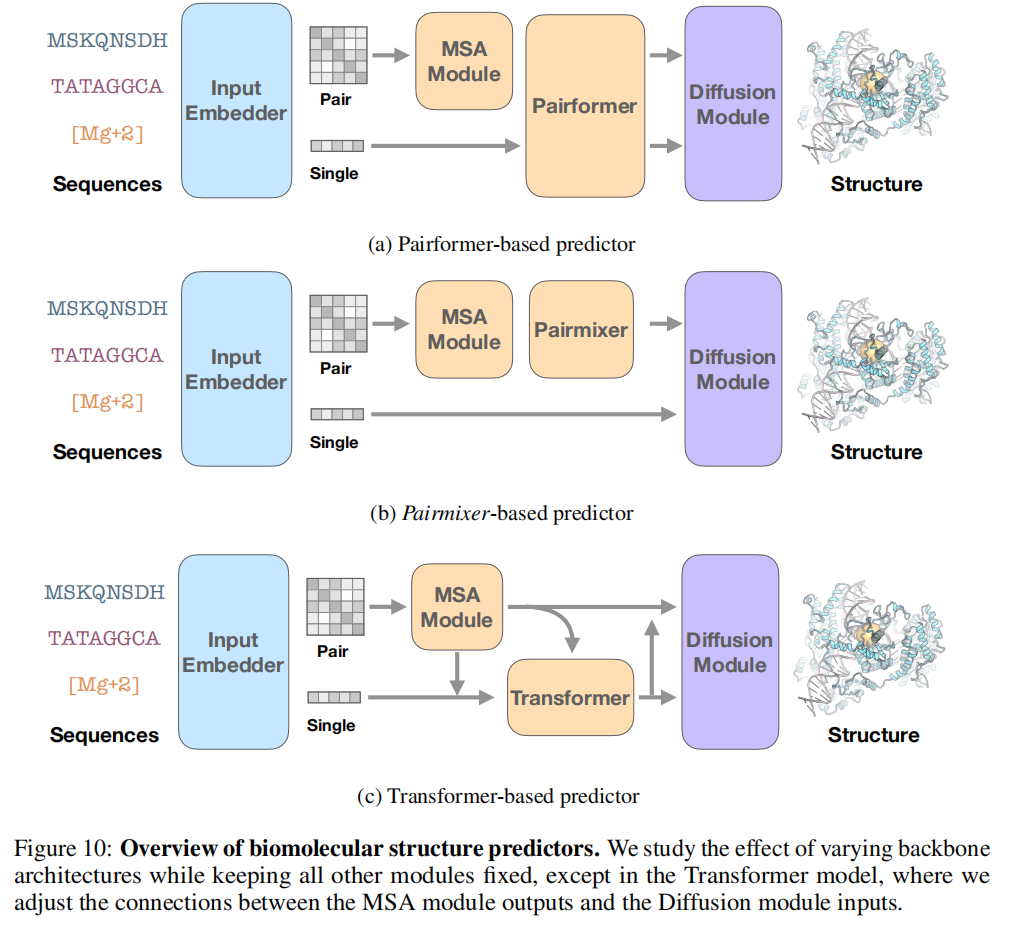
2.1 AlphaFold3 的整体流程
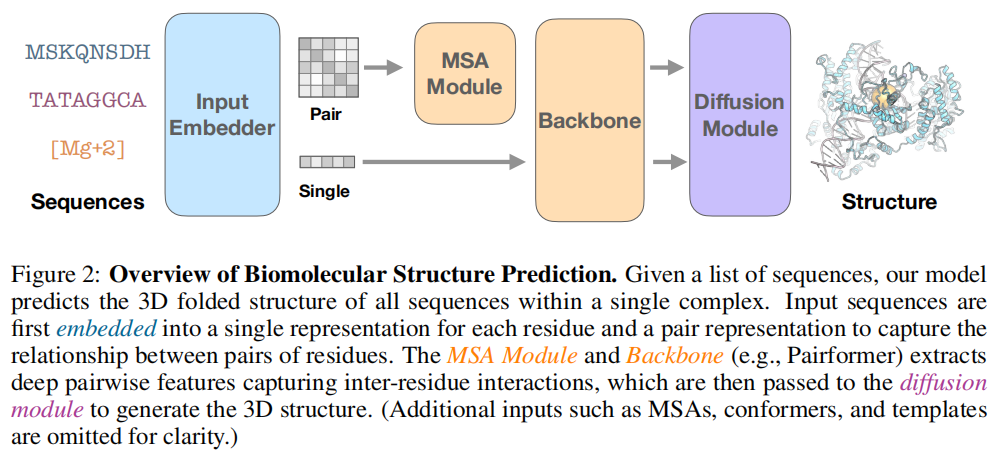
AlphaFold3 等的推理流程可以分为四个阶段:
输入序列 (蛋白质/核酸/小分子)
↓
Input Embedder(初始化单序列表示 s^init 和配对表示 z^init)
↓
MSA Module(从多序列比对中提取共进化特征,输出 z^msa)
↓
Pairformer Backbone(深层特征提取,输出 s^backbone, z^backbone)
↓
Diffusion Module(扩散去噪,生成全原子三维坐标)其中,Pairformer 是计算的核心瓶颈,也是本文的主要研究对象。
2.2 Pairformer 的四大组件
(1)三角注意力(Triangle Attention)
对配对矩阵的第 行做带配对偏置的注意力:
其中 是由完整配对表示投影出的注意力偏置项。直觉上, 在以残基 为"锚点"的条件下,对所有残基对进行注意力聚合,实现残基三元组(triplet)层面的几何推理。
实践中需要分别对行和列各做一次,总 FLOPs 为 。
(2)三角乘法(Triangle Multiplication)
对每条边 ,通过将其与所有"中间节点" 的边特征做 Hadamard 乘积并求和,隐式地对残基三元组 建模。以 torch.einsum 实现,FLOPs 为 ,但因矩阵乘法的高度优化,实际效率远优于三角注意力。
(3)序列更新(Sequence Update)
先做带配对偏置的序列注意力,再接前馈网络:
(4)前馈网络(FFN)
作用于配对表示中的每一个位置对,是标准的逐位置变换。
三、方法:Pairmixer 的设计哲学
3.1 核心假设与移除依据
移除三角注意力的依据:
三角注意力和三角乘法都能实现对残基三元组的推理,本质上是同一类能力的两种实现。前者通过稀疏的选择性聚合实现,后者通过密集的矩阵乘法实现。作者认为,在充分训练后,三角乘法完全可以通过调整配对表示的数值幅度来隐式模拟稀疏选择——密集操作并不意味着无法学到稀疏的有效交互(这一点在第六节分析中有实验验证)。
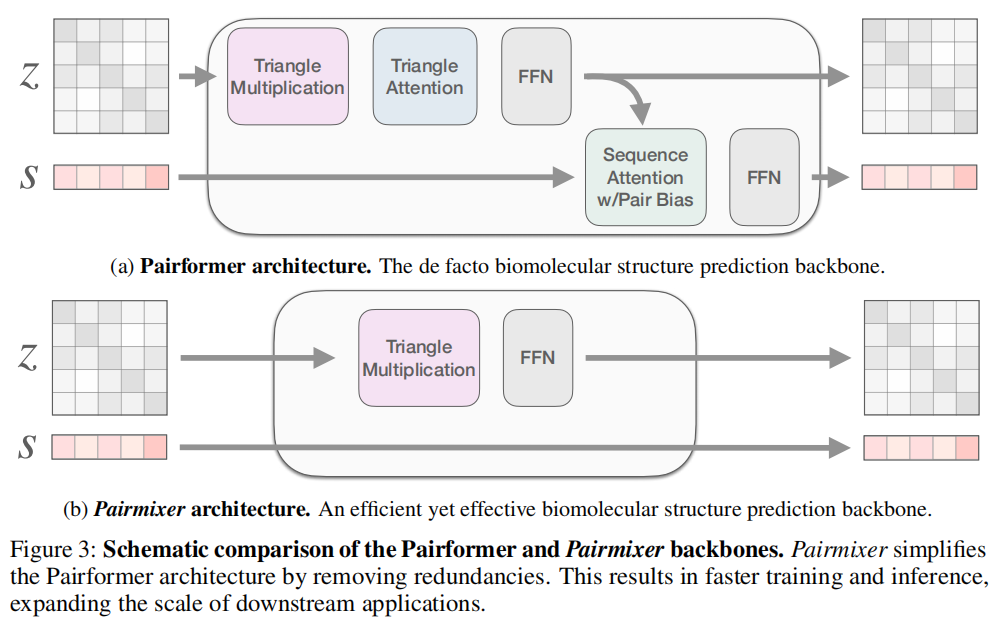
移除序列更新的依据:
在 AlphaFold2 的 Evoformer 中,序列更新是从 MSA 中提取进化特征的关键路径。但在 AlphaFold3 的流程中,MSA Module 已经承担了这一职责,其输出 包含了足够的共进化信息。将此信息再次通过骨干网络的序列更新路径传递,实属冗余。因此,Pairmixer 直接将 绕过骨干网络传给扩散模块()。
3.2 Pairmixer 算法
最终的 Pairmixer 极度简洁,完整算法如下:
输入:配对表示 z^msa ∈ R^{L×L×Cz},骨干层数 N
输出:更新后的配对表示 z^N
z^0 ← z^msa
for l = 0 to N-1:
z^l ← z^l + TriMulIncoming(z^l) # 按行的三角乘法
z^l ← z^l + TriMulOutgoing(z^l) # 按列的三角乘法
z^{l+1} ← z^l + FFN(z^l)
return z^N与 Pairformer 相比,Pairmixer 去除了三角注意力和序列更新两条路径,只保留了:
- • 双向三角乘法(入向 + 出向)
- • 逐位对前馈网络
3.3 计算复杂度对比
模块 | FLOPs(主项) | 主要瓶颈 |
|---|---|---|
三角注意力(行+列) | 次独立注意力计算 | |
三角乘法(行+列) | einsum,可高度并行 | |
序列注意力(带配对偏置) | 二次项,影响相对较小 | |
FFN(配对) | 二次项 |
Pairmixer 在每层内消除了 的三角注意力开销,从而实现整体加速。
四、实验设置
4.1 实现细节
研究团队将 Pairmixer 实现于 Boltz-1 之上,仅替换 Pairformer 骨干和 MSA Module 中的三角注意力,扩散模块保持不变。同时设置了一个纯序列 Transformer 基线(去掉配对更新,保留序列更新),以厘清配对表示本身的贡献。
训练遵循 Boltz-1 的调度策略:
- • 第一阶段:53k 步,使用 PDB + OpenFold 蒸馏数据,token/atom crop 为 384/3456
- • 第二阶段(微调):15k 步,仅使用 PDB 数据,crop 扩大至 512/4608
- • 推理时默认 10 次 recycling、200 个扩散采样步
4.2 模型规模
规模 | 骨干层数 | 扩散层数 | 对应基准 |
|---|---|---|---|
Small | 12 | 6 | — |
Medium | 24 | 24 | — |
Large | 48 | 24 | Boltz-1 同规模 |
4.3 评测集
- • RCSB 测试集(533 个结构):序列一致性 ≤40%,小分子相似度 ≤80%,分辨率 <4.5Å
- • CASP15 测试集(66 个结构):国际蛋白质结构预测竞赛标准集
- • PoseBusters 基准(298 个蛋白质-配体复合物)
- • 抗体-抗原复合物数据集(70 个,来自 AlphaFold3 论文)
- • 蛋白质-核酸复合物 + RNA 结构(172 + 27 个,来自 AlphaFold3 论文)
五、结果分析
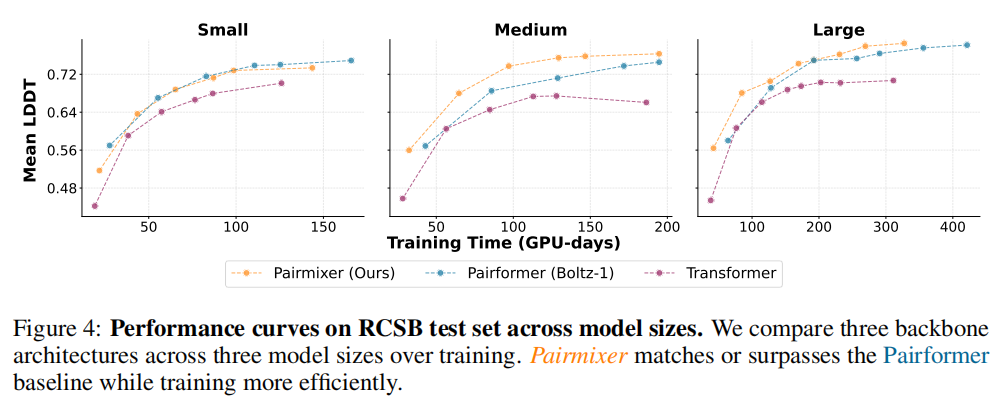
5.1 跨规模训练效率
在 RCSB 测试集的 Mean LDDT 指标上:
- • Large 规模:Pairmixer 用 66% 的训练时间(192 vs 290 GPU-days)达到与 Pairformer 相同的 Mean LDDT(0.78)
- • Medium 规模:相同训练时间下,Pairmixer 超越 Pairformer
- • Small 规模:与 Pairformer 持平
- • 在所有规模下,Pairmixer 均显著优于纯序列 Transformer 基线
这一结果表明:序列 Transformer 对结构特征的表达能力存在根本性缺陷,而三角乘法 + FFN 已经足以捕捉丰富的结构表示。
5.2 推理速度
在 GH200 GPU、512 token 默认配置下:
序列长度 | Boltz-1 (Pairformer) | Pairmixer | 加速比 |
|---|---|---|---|
512 tokens | 34 秒 | 21 秒 | 1.6× |
1024 tokens | ~200 秒 | ~100 秒 | 2× |
2048 tokens | 1000 秒 | 250 秒 | 4× |
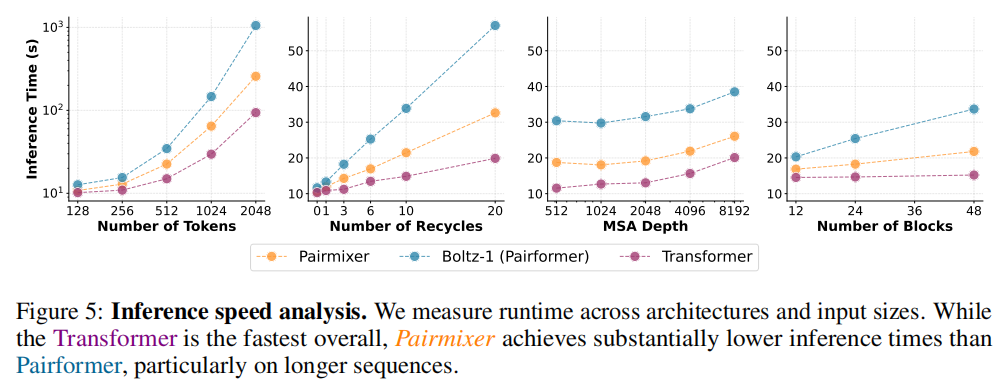
加速比随序列长度非线性增长,这与三角注意力的 复杂度高于三角乘法的规律一致。
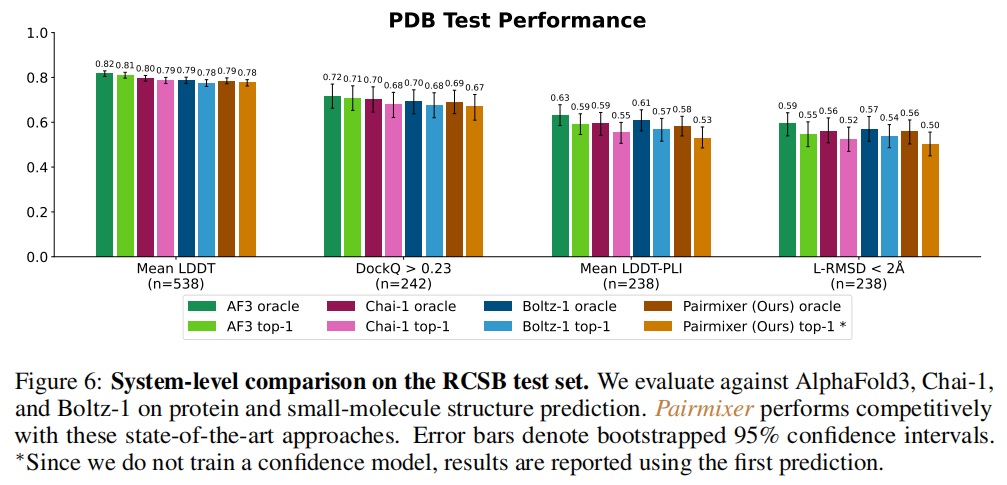
5.3 与 SOTA 模型的系统对比(RCSB 测试集)
方法 | Mean LDDT | DockQ>0.23 | Mean LDDT-PLI | L-RMSD<2Å |
|---|---|---|---|---|
AlphaFold3 (oracle) | 0.82 | 0.72 | 0.63 | 0.59 |
Chai-1 (oracle) | 0.80 | 0.70 | 0.59 | 0.56 |
Boltz-1 (oracle) | 0.79 | 0.68 | 0.55 | 0.52 |
Pairmixer (oracle) | 0.79 | 0.68 | 0.57 | 0.54 |
Pairmixer 与 Boltz-1 持平,在 LDDT-PLI 上甚至略有提升(0.57 vs 0.55),与 AF3 的差距主要体现在蛋白质-蛋白质对接(DockQ)上。
5.4 多样化生物分子任务
任务 | 指标 | Pairformer (Boltz-1) | Pairmixer | Transformer |
|---|---|---|---|---|
蛋白质-配体 (PoseBusters) | RMSD<2Å | 0.68 | 0.67 | — |
抗体-抗原 | DockQ>0.23 | 0.23 | 0.23 | 0.08 |
蛋白质-核酸 | IPS | 0.65 | 0.66 | 0.64 |
RNA 结构 | lDDT | 0.58 | 0.59 | 0.61 |
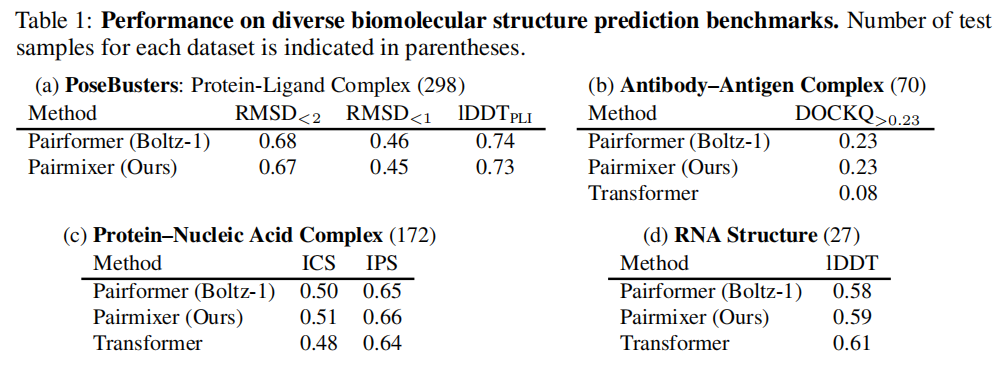
值得注意的是,在 RNA 结构预测上,Transformer 基线略优,作者认为这与 RNA 训练数据稀少、配对表示的归纳偏置反而不利于过拟合保护有关。
5.5 BindFast:Pairmixer 赋能蛋白质设计
将 Pairmixer 嵌入 BoltzDesign 框架,形成 BindFast:
目标蛋白 | 序列总长 | Pairformer 耗时 | Pairmixer 耗时 | 加速比 |
|---|---|---|---|---|
GIP peptide (2QHK_B) | 140 | 680s | 337s | 2.01× |
Ubiquitin (1UBQ_A) | 186 | 1113s | 532s | 2.09× |
TP53 (4MZI_A) | 303 | 3198s | 1390s | 2.30× |
hSDH (1P5J_A) | 429 | 7289s | 2920s | 2.50× |
hMAO (1GOS_A) | 607 | 17134s | 6601s | 2.60× |
bsDNA Polymerase (3TAN_A) | 702 | OOM | 9184s | ∞ |
hTLR3 (1ZIW_A) | 739 | OOM | 10568s | ∞ |
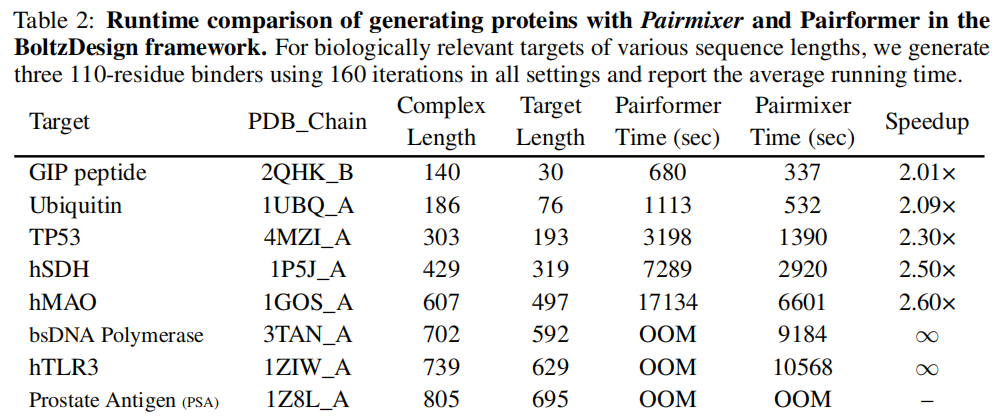
Pairmixer 将可设计目标的序列上限从 500 残基扩展至 650 残基(提升 30%),并在可运行范围内实现 2x–2.6x 加速。
六、深度分析:三角乘法为何"够用"
6.1 配对表示的核心价值
通过将 Pairmixer 与纯序列 Transformer 基线的逐样本对比:
指标 | Pairmixer 胜率 | 解读 |
|---|---|---|
lDDT(基于局部距离) | 93.7% | 配对表示对残基间精细空间关系的捕捉能力 |
RMSD(需全局对齐) | 73.3% | 全局构象在两种架构间差异较小 |
DockQ | 71.6% | 接口精度同样受益于配对表示 |
lDDT-PLI | 64.8% | 蛋白质-配体交互同样获益 |
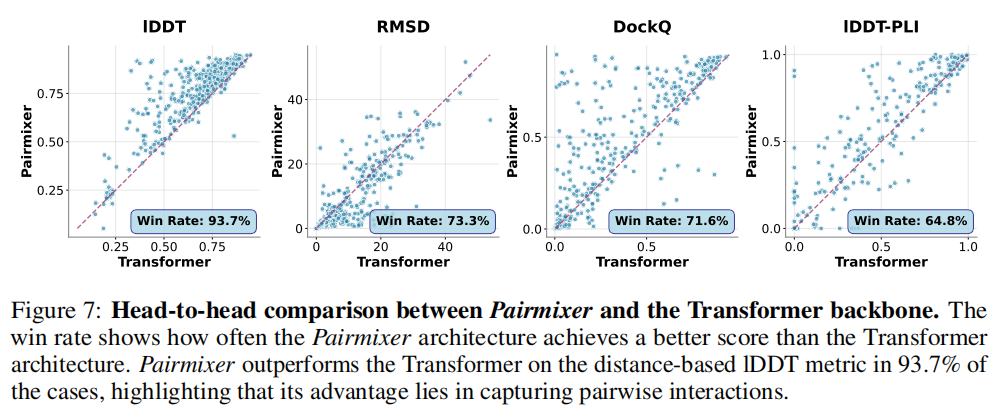
lDDT 胜率高达 93.7% 而 RMSD 胜率仅 73.3% 的对比,精确揭示了配对表示的贡献来源:局部残基间距离的精确建模,而非全局折叠形状。
6.2 三角乘法的稀疏性分析
为验证三角乘法是否真的在密集计算中学到了稀疏交互,研究者设计了两种推理时 dropout 方案:
随机 dropout(Random Dropout):以概率 随机屏蔽三角乘法中的项。结果显示, 时性能急剧下降。
低范数 dropout(Low-Norm Dropout):屏蔽范数最小的 比例的项(即"最不活跃"的交互)。结果显示,屏蔽高达 75% 的低范数项后,性能几乎不变。
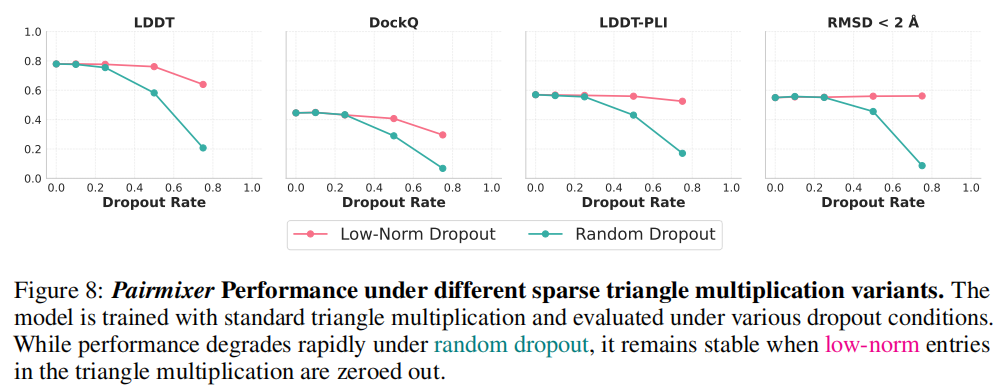
这两个实验共同证明:三角乘法自发地将计算能量集中在少数高范数的关键交互上,从而实现了与注意力机制类似的稀疏特性,但无需显式的 softmax 选择机制。
6.3 局部性假设的破伪
进一步使用块状 dropout(blockwise dropout)测试局部交互的充分性:只保留 的"局部"交互项。结果显示:
- • 时性能开始下降
- • 时出现显著损失
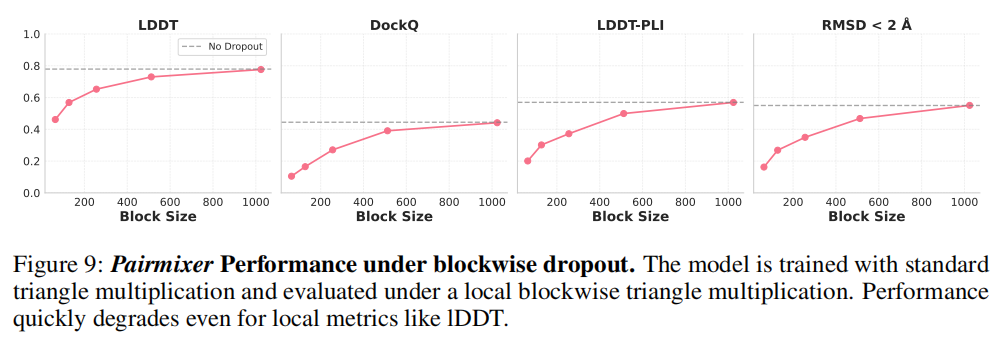
这排除了"三角乘法只依赖局部接触"的假设,证实其捕捉的是真正的稀疏长程交互(sparse long-range interactions),在分子间界面和三维空间中距离近但序列距离远的残基对上尤为关键。
6.4 Pairformer 消融实验
在 Small 规模的 Pairformer 上逐模块消融:
消融项 | GPU-days | lDDT | DockQ>0.23 | lDDTPLI |
|---|---|---|---|---|
完整 Pairformer | 82 | 0.74 | 0.57 | 0.52 |
去掉序列更新 | 80 | 0.73 | 0.57 | 0.54 |
去掉三角注意力 | 66 | 0.70 | 0.55 | 0.50 |
去掉三角乘法 | 71 | 0.70 | 0.53 | 0.49 |
在短训练周期(60 epochs)下,三角注意力和三角乘法都有贡献;但作者指出,Pairmixer 在更长训练后可以恢复并超越这一基线,说明三角乘法在充分训练时能弥补注意力的缺失。
6.5 Match3 基准:配对表示 vs. 三次方计算的独立验证
为隔离配对表示和三次方计算各自的贡献,论文在 Match3 任务(给定序列,判断是否存在三个元素之和对 M 取模为零的三元组)上进行对比:
架构 | 表示维度 | 计算量级 |
|---|---|---|
标准自注意力 | 线性(1D) | |
三阶自注意力 | 线性(1D) | |
三角乘法 | 二次(2D 配对) |
结果:当数据充足且模型加深时,三角乘法优于两种注意力变体,证明配对表示带来的归纳偏置(而非仅仅三次方计算量)是关键优势。
七、相关工作与定位
工作 | 特点 | 与 Pairmixer 的区别 |
|---|---|---|
AlphaFold2 Evoformer | 三角注意力 + 三角乘法 + MSA 更新 | 针对单体折叠,计算代价更高 |
AlphaFold3 Pairformer | 去 MSA 更新,加三角操作 | 仍含三角注意力 |
MiniFold (2025) | 用三角乘法简化 Evoformer | 仅针对单体折叠(AlphaFold2 风格) |
SimpleFold (2025) | 纯序列 Transformer,去掉配对表示 | 本文实验证明这是有损的简化 |
MSA Pairformer (2025) | 将三角操作用于 MSA 特征提取 | 不针对结构预测骨干 |
Pairmixer(本文) | 三角乘法 + FFN,无注意力 | AlphaFold3 风格的共折叠模型,覆盖全模态 |
Pairmixer 的独特之处在于:它是首个在 AlphaFold3 风格的多模态共折叠(cofolding)框架中,系统验证"三角乘法 + FFN 足以替代全部注意力机制"的工作。
八、局限性与未来方向
- 1. 训练规模上限:实验中的大规模配置与 Boltz-1 对齐,但与 AlphaFold3 完整训练规模(数十亿参数、超大数据集)仍有差距,CASP15 等小测试集上的评估存在较大方差。
- 2. RNA 预测略弱:在 RNA-only 结构上,纯序列 Transformer 反而略优于 Pairmixer,说明对于训练数据极度稀缺的模态,配对表示的归纳偏置并非总是有益。
- 3. 置信度模型缺失:当前 Pairmixer 没有训练专用的置信度模型,系统评估中只能使用第一个预测样本,而非置信度排序后的最优样本,这在公平对比上存在一定劣势。
- 4. 稀疏化推理:低范数 dropout 实验表明 75% 的交互是低信息量的,未来可探索在训练时引入稀疏化约束,进一步压缩三角乘法的实际计算量(从 向亚三次方迈进)。
- 5. 更大蛋白质复合物:BindFast 已将可设计目标延伸至 ~650 残基,但人类蛋白质组中仍存在大量更长的靶标,突破这一上限仍是工程挑战。
九、总结与启示
Pairmixer 的核心贡献可以归纳为三点:
1. 方法论贡献:通过严格的消融实验证明,在 AlphaFold3 风格的共折叠模型中,三角注意力和序列更新均是冗余的;三角乘法 + FFN 是使配对表示发挥作用的最小充分条件。
2. 工程贡献:在不降低精度的前提下,实现了 1.5× 训练加速和长序列上的 4× 推理加速,将大规模结构预测应用的可行性边界显著扩展。
3. 理论洞见:通过稀疏 dropout 实验,揭示了三角乘法在密集计算形式下隐式实现稀疏长程几何推理的能力,为理解为何"二维配对表示"对结构预测如此关键提供了新的证据。
对于从事计算生物学、AI for Science、以及深度学习架构研究的读者而言,这篇工作提供了一个令人信服的案例:在领域专用模型中,有针对性地去除冗余模块,往往比盲目引入新组件更有价值
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-10,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号