复杂相关性散点图复现(ggplot2绘图的层层递进)


今天给大家复现的图来自文献《Epigenetic age acceleration of cervical squamous cell carcinoma converged to human papillomavirus 16/18 expression, immunoactivation, and favourable prognosis》

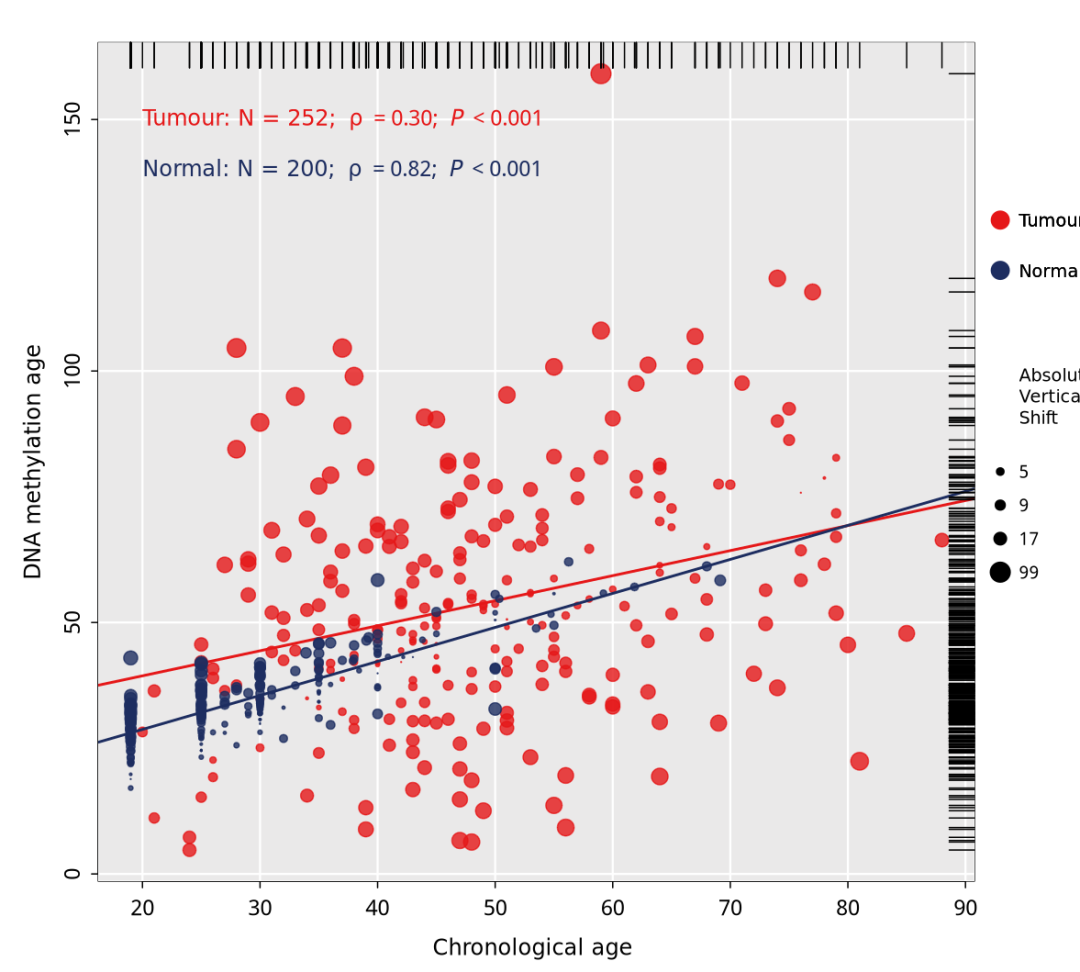
原文的图如下所示,现在我们开始复现里面的a图,可以很清晰的看到
在200个正常样本中,实际年龄与DNAm年龄高度相关(ρ = 0.82)。然而,这种相关性在肿瘤组织中很大程度上缺失(ρ=0.30),这表明在正常宫颈组织中观察到的DNA甲基化模式在宫颈肿瘤组织中被破坏( P <0.001;图1a )
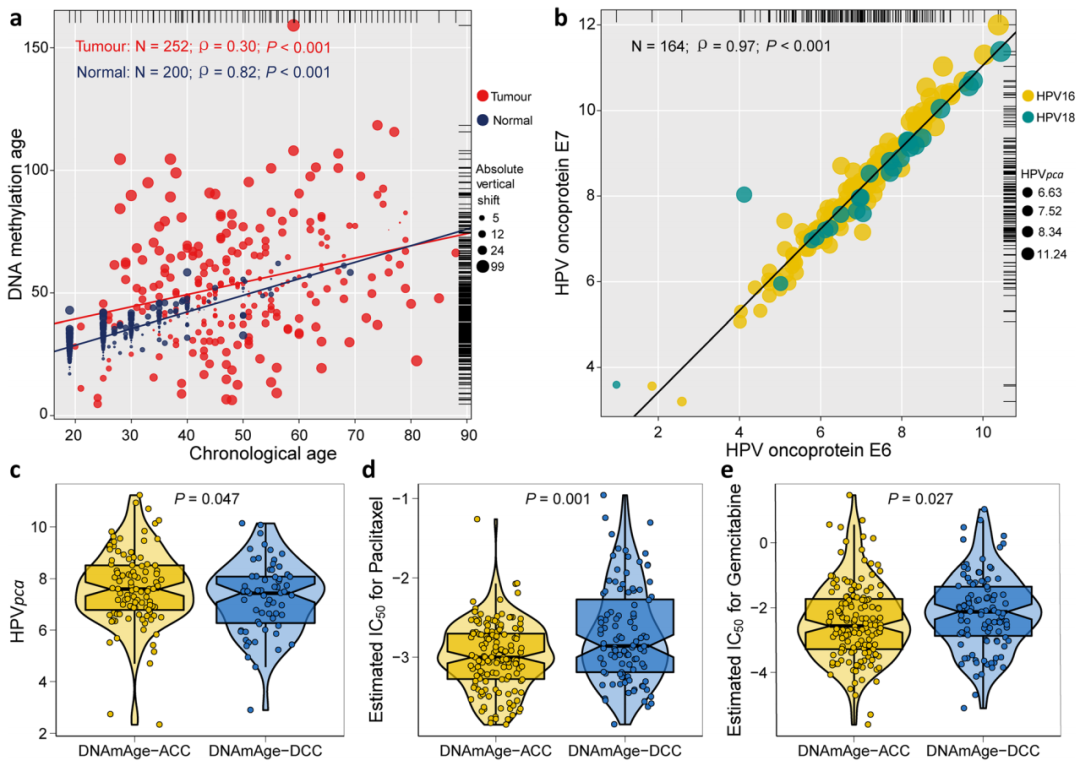
Fig. 1 Correlations between DNAm age and chronological age and other molecular characteristics of DNAm age groups
数据队列
作者收集了来自TGCA 的252个宫颈鳞状细胞癌样本,TCGA与GEO的 200 个正常组织样本。Fig1 展示了 在正常组织和肿瘤组织中,DNA甲基化年龄与实际年龄之间的相关性存在差异。
DNA甲基化年龄的计算方法如下:
DNA methylation age, based on Horvath’s clock model, was calculated from the methylation β values using the agep() embedded in R package “wateRmelon”
生理年龄:就是样本的实际年龄
准备数据
## 读取数据
data <- read.csv("input.csv")
head(data)
table(data$class)

先计算散点图里点的大小
样本根据DNA甲基化年龄与实际年龄之间的差异即差值进一步被分为两类:表观遗传年龄加速组 ‘DNAmAge-ACC’ 或年龄减缓组 ‘DNAmAge-DEC’。如果差值大于零,则被归类为 “DNAmAge-ACC” 或年龄加速组;如果偏移小于零,则被归类为“DNAmAge-DEC”或年龄减缓组。
# 计算甲基化加速年龄
data$diff <- abs(data$dnamage - data$age)
# 根据加速程度计算散点大小,对diff值进行log转换
data$size <- log10(data$diff + 1)
# 计算图例里点的大小,分配散点大小区间
# quantile(data$size)返回四分位数,根据四分位数将数据划分为4个区间
data$range <- cut(data$size, breaks = quantile(data$size), include.lowest = T)
# 取区间的后半部分,用于绘制图例
data$range2 <- as.numeric(gsub("]", "", sapply(strsplit(as.character(data$range),","), "[",2), fixed = T))
head(data)

计算图中左上角的相关性
Tumor组中的DNAm Age(DNA甲基化年龄)与Chronological Age(生理年龄)相关性:
# Tumor组中的DNAm Age(DNA甲基化年龄)与Chronological Age(生理年龄)相关性
data_t <- data[data$class == "T", ]
cor_t <- cor.test(data_t$age, data_t$dnamage)
cor_t
# Pearson's product-moment correlation
#
# data: data_t$age and data_t$dnamage
# t = 4.914, df = 250, p-value = 1.615e-06
# alternative hypothesis: true correlation is not equal to 0
# 95 percent confidence interval:
# 0.1798074 0.4054876
# sample estimates:
# cor
# 0.2967859
Normal 组中的DNAm Age(DNA甲基化年龄)与Chronological Age(生理年龄)相关性:
# Normal 组中的DNAm Age(DNA甲基化年龄)与Chronological Age(生理年龄)相关性
data_n <- cor.data[data$class == "N", ]
cor_n <- cor.test(data_n$age, data_n$dnamage)
cor_n
# Pearson's product-moment correlation
#
# data: data_n$age and data_n$dnamage
# t = 20.296, df = 198, p-value < 2.2e-16
# alternative hypothesis: true correlation is not equal to 0
# 95 percent confidence interval:
# 0.7709783 0.8622419
# sample estimates:
# cor
# 0.821813
绘图
1、使用 par,plot,rect,grid基础函数 对画布进行基本设置
# 画布基本设置
par(bty="o", mgp = c(2,0.5,0), mar = c(4.1,4.1,2.1,4.1), tcl=-.25, font.main=3)
# 先绘制一个空的画布,仅有边框和坐标名
plot(NULL, NULL, ylim = ylim, xlim = xlim, xlab = "Chronological age ", ylab = "DNA methylation age",col="white",main = "")
# rect基础函数 给画布设置背景色,掩盖边框
rect(par("usr")[1], par("usr")[3], par("usr")[2], par("usr")[4], col = "#EAE9E9",border = F)
# grid函数添加网格
grid(col = "white", lty = 1, lwd = 1.5)
得到如下:
2、画散点和回归线
# 在画布中添加肿瘤组的散点
points(data_t$age, data_t$dnamage, pch = 19, col = ggplot2::alpha("#E51718",0.8),cex = data_t$size)
# 添加回归线
abline(lm(dnamage~age, data=data_t), lwd = 2, col = "#E51718")
# 在画布中添加正常组的散点
points(data_n$age, data_n$dnamage, pch = 19, col = ggplot2::alpha("#1D2D60",0.8), cex = data_n$size)
abline(lm(dnamage~age, data=data_n), lwd = 2, col = "#1D2D60")
结果如下:
3、画顶部和右侧地毯线
# 添加边际地毯线显示数据分布情况
rug(data$age, col="black", lwd=1, side=3)
rug(data$dnamage, col="black", lwd=1, side=4)
4、添加相关性结果
text(20,150, adj = 0,expression("Tumour: N = 252; "~rho~" = 0.30; "~italic(P)~" < 0.001"), col = c("#E51718"), cex=1)
text(20,140,adj = 0,expression("Normal: N = 200; "~rho~" = 0.82; "~italic(P)~" < 0.001"), col = c("#1D2D60"), cex=1)
5、画图例
# 计算图例里需要绘制多少圆圈
num <- length(unique(data$range2))
num
# 散点图例
points(x = rep(par("usr")[2] + 2.2, num), y = seq(80,60, length.out = num),
pch = 19, bty = "n", cex = sort(unique(data$range2)), col = "black")
# 点的文字
text(x = rep(par("usr")[2] + 3.8, num + 1), y = c(95, seq(80,60,length.out = num)),
labels = c("Absolute\nVertical\nShift", round(10^(sort(unique(data$range2))) - 1,0)),
adj = 0,cex = 0.8)
# 做分组的圆圈(肿瘤和正常)
points(x = rep(par("usr")[2] + 2.2, 2), y = c(130, 120),
pch = 19, bty = "n", cex = 1.8, col = c("#E51718","#1D2D60"))
# 做分组图图例的文字
text(x = rep(par("usr")[2] + 3.8, num + 1), y = c(130, 120),
labels = c("Tumour","Normal"), adj = 0,cex = 0.8)
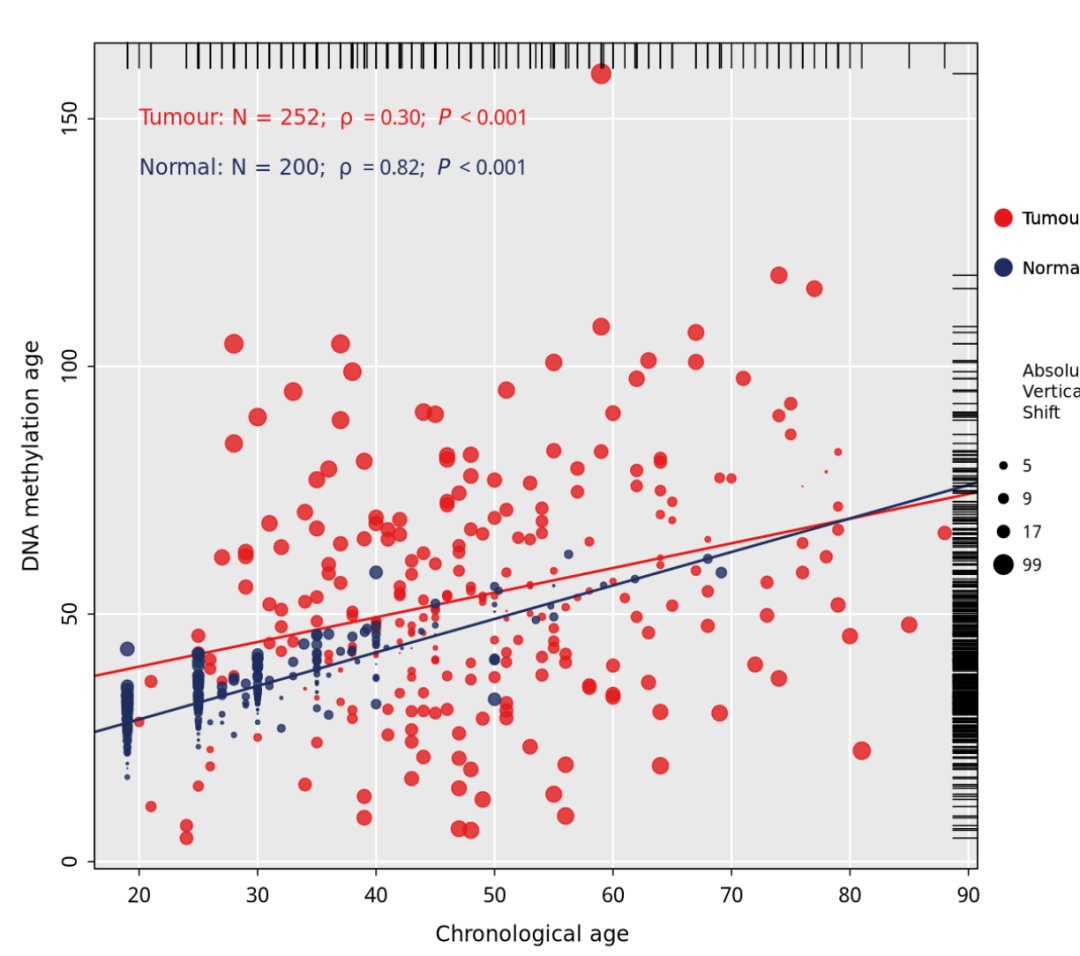
6、添加边框
# 设置new = TRUE时,新的图形会叠加在现有的图形上
# 设置bty="o"会使得图形具有一个完整的矩形边框
par(new = T, bty="o")
# 这行代码创建一个空白的图形窗口,具有指定的坐标轴范围,但没有轴标签和刻度。
ylim <- range(data$dnamage)
xlim <- range(data$age)
plot(-1, -1, col = "white",xlim = xlim, ylim = ylim, xlab = "", ylab = "", xaxt = "n", yaxt = "n")
最终结果如下:
最后
如果你要从ggplot2开始一步步调制成为它这样的美图,需要下很深的功夫,一张统计图就是从数据到几何对象(点、线、条形等)的图形属性(颜色、形状、大小等)的一个映射。
- ✦ 数据(Data),最基础的是可视化的数据和一系列图形映射(aesthetic mappings),该映射描述了数据中的变量如何映射到可见的图形属性。
- ✦ 几何对象(Geometric objects, geoms)代表在图中实际看到的点、线、多边形等。
- ✦ 统计转换(Statistical trassformations, stats)是对数据进行某种汇总,例如将数据分组创建直方图,或将一个二维的关系用线性模型进行解释。
- ✦ 标度(Scales)是将数据的取值映射到图形空间,例如用颜色、大小或形状来表示不同的取值,展现标度的常见做法是绘制图例和坐标轴。
- ✦ 坐标系(Coordinate system, coord)描述数据是如何映射到图形所在的平面,同时提供看图所需的坐标轴和网格线。
- ✦ 分面(faceting)如何将数据分解为子集,以及如何对子集作图并展示。
- ✦ 主题(theme)控制细节显示,例如字体大小和图形的背景色。
基础绘图永远是基本功,我们之前讲过一个公开课,欢迎观看:https://www.bilibili.com/video/BV1Wi4y1A7u5/

分组后做转录组差异分析
根据 DNAm 年龄和实际年龄之间的变化,将患者进一步分为表观遗传年龄加速组或年龄减速组。
- 如果偏移大于零,它们被分类为“DNAmAge-ACC”或年龄加速;
- 如果偏移小于零,则它们被分类为“DNAmAge-DEC”或年龄减速。
文章里面就做了这个分组后的转录组崔西,大家可以试试看,作为学徒作业哈;
we performed differential expression analysis and identified 103 and 184 significantly upregulated genes (fold change > 2, false discovery rate (FDR) < 0.05) for the DNAm-ACC and DNAm-DEC groups, respectively
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-01-05,如有侵权请联系 cloudcommunity@tencent.com 删除
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号