Nucleic Acids Res. | GPSFun:使用语言模型的几何感知蛋白序列功能预测

DRUGAI
今天为大家介绍的是来自中山大学杨跃东团队的一篇论文。了解蛋白质功能对于阐明疾病机制和发现新药靶点至关重要。然而,蛋白质序列的指数增长与其有限的功能注释之间的差距正在扩大。在之前的研究中,作者开发了一系列方法,包括GraphPPIS、GraphSite、LMetalSite和SPROF-GO,用于蛋白质残基或蛋白质水平的功能注释。为了进一步提高这些方法的适用性和性能,作者现推出GPSFun,这是一款用于几何感知蛋白质序列功能注释的多功能网络服务器,结合了语言模型和几何深度学习以提升以往工具的性能。具体而言,GPSFun利用大型语言模型高效预测输入蛋白质序列的3D构象,并提取有用的序列嵌入。随后,几何图神经网络被用来捕捉蛋白质图中的序列和结构模式,从而促进各种下游预测,包括蛋白质-配体结合位点、基因本体论(gene ontologies)、亚细胞位置(subcellular locations)和蛋白质溶解度(protein solubility)。值得注意的是,GPSFun在各种任务中均表现优于最新的前沿方法,不需要多序列比对或实验蛋白质结构。GPSFun对所有用户免费开放,并提供用户友好的界面和丰富的可视化功能,网址为https://bio-web1.nscc-gz.cn/app/GPSFun。

了解蛋白质功能对于理解宏基因组功能、揭示疾病机制和发现新药靶点至关重要。由于生化实验确定蛋白质功能的过程昂贵、耗时且通量低,目前蛋白质序列的迅速扩展与其有限的功能注释之间的差距正在扩大。为此,已经开发了许多计算工具,用于蛋白质残基和蛋白质水平的功能预测,例如蛋白质-配体结合位点、基因本体论(GO)、亚细胞位置和蛋白质溶解度。
尽管有大量针对各种任务设计的蛋白质功能预测工具,但缺乏一个提供高质量预测的全面平台。此外,许多现有的基于序列的方法,如TargetS,严重依赖于多序列比对(MSA),这在计算上非常昂贵,对于缺乏近亲同源物的孤儿蛋白来说是无效的。虽然作者之前的研究,如LMetalSite和SPROF-GO,通过用语言模型表示替代MSA克服了这个问题,但结构信息的缺失仍然提供了提高准确性的机会。相比之下,通过图神经网络(GNN)编码蛋白质结构的实验结构方法通常更有效。然而,这些方法中的大多数尚未充分探索结构中的几何形态。更重要的是,基于结构的方法不适用于尚未解决结构的新蛋白质。尽管作者之前开发的GraphSite已经展示了利用AlphaFold2预测结构进行DNA结合位点预测的可行性,但计算密集的结构预测流程限制了其在AlphaFold蛋白质结构数据库中缺失的序列上的应用。
基于蛋白质语言模型的最新发展,ESMFold作为AlphaFold2的一个有前途的替代方案,它用大规模预训练的蛋白质语言模型替代MSA,大大加快了预测速度,同时保持了相当的准确性。为了促进蛋白质结构建模,几何深度学习最近在蛋白质结构预训练、蛋白质设计、蛋白质对接和结合位点预测方面蓬勃发展。基于这些最新进展,有望进一步提高作者之前验证良好的蛋白质功能注释方法的适用性和性能。
模型部分
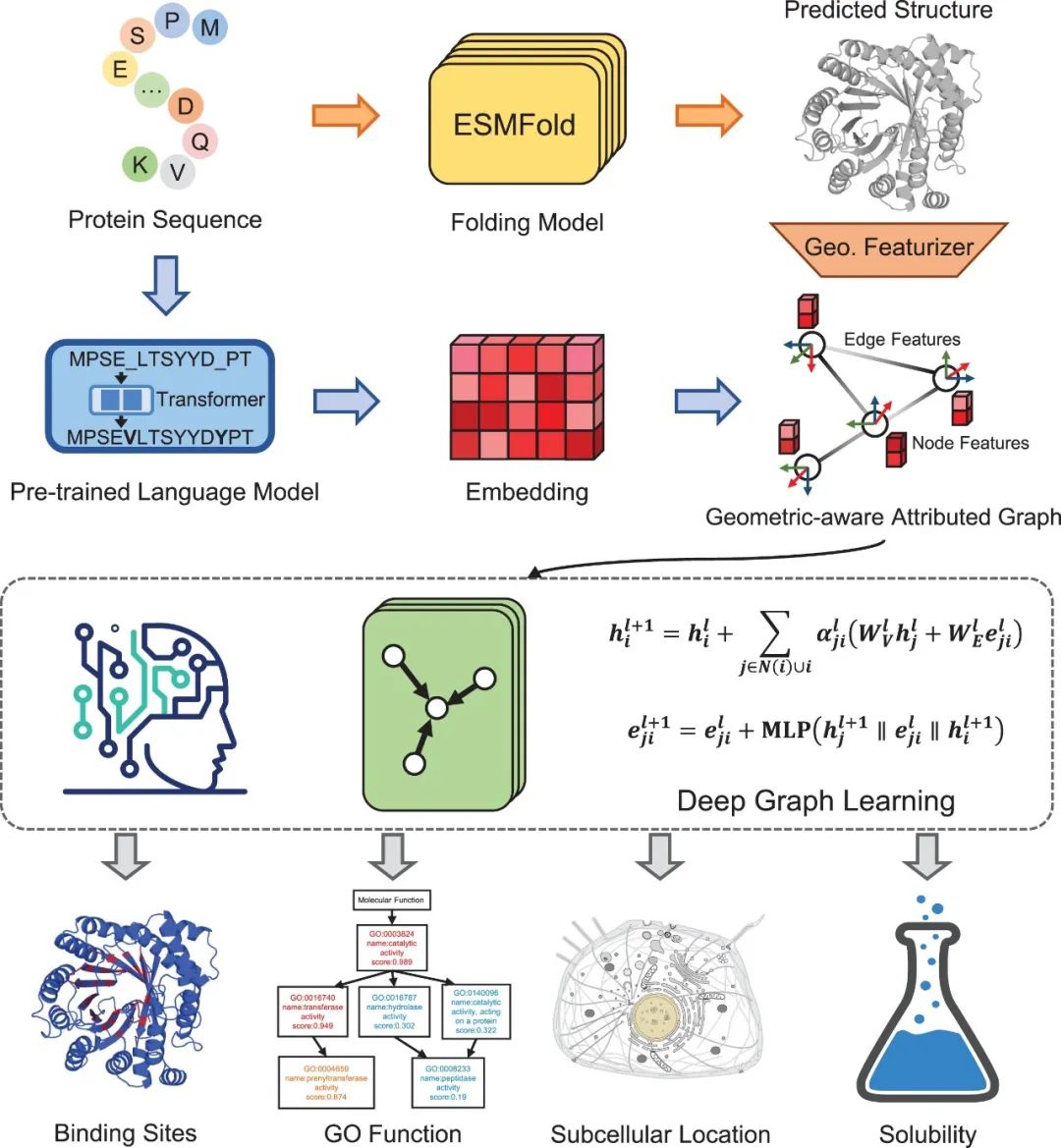
图 1
GPSFun的工作流程如图1所示。对于输入序列,GPSFun首先采用基于语言模型的折叠算法ESMFold预测蛋白质的3D构象。然后,使用另一个预训练的蛋白质语言模型ProtTrans(版本:ProtT5-XL-U50)提取序列嵌入,并通过最小-最大归一化进行进一步标准化。随后,使用几何特征提取器(geometric featurizer)捕捉预测结构中的残基和关系几何上下文。作者还使用DSSP从预测结构中计算相对溶剂可及性和二级结构谱,就像之前的研究中所做的一样。生成的几何感知蛋白质属性图被输入到一组GNN中,以发现用于各种下游任务(包括蛋白质-配体结合位点、GO功能、亚细胞定位和溶解度预测)的高级模式。
对于几何特征提取器,GPSFun将蛋白质表示为一个半径图,其中残基构成节点,相邻节点(Cα之间的距离小于15 Å)通过边连接。使用端到端的特征提取器来提取几何特征,类似于之前的方法,不同之处在于作者还对残基的侧链构象进行编码。具体而言,首先基于骨架Cα、N和C原子的相对位置在每个残基处定义一个局部坐标系。然后,导出几个SE(3)-不变的几何特征,以捕捉残基内或残基间骨架和侧链原子的排列情况。几何节点特征包括任何两个原子之间的残基内距离、其他内原子相对于Cα的相对方向,以及键(bond)和扭转角(torsion angles)。几何边特征包括相邻残基中任何两个原子之间的残基间距离、相邻残基中所有原子相对于中心残基Cα的相对方向,以及相邻节点两个参考系之间的旋转角。为了编码侧链构象,作者计算了重侧链原子的质心,并作为常规原子参与上述特征计算。
实验设置
为了训练蛋白质-配体结合位点、亚细胞定位和溶解度预测模型,作者在训练集上进行了五折交叉验证。对于GO预测,模型使用五个不同的随机种子在训练集上进行训练,并在预定义的验证集上进行评估。所有超参数通过网格搜索基于验证集的性能进行优化。在测试阶段,使用所有五个训练模型(来自交叉验证或不同种子)进行预测,并将其平均作为GPSFun的最终预测结果。
作者使用召回率(Rec)、精度(Pre)、准确率(Acc)、Jaccard系数、F1得分(F1)、最大蛋白质中心F值(Fmax)、Matthew相关系数(MCC)、受试者工作特征曲线下面积(AUC)和精确率-召回率曲线下面积(AUPR)来评估预测性能。
GPSFun在蛋白质-配体结合位点上的性能
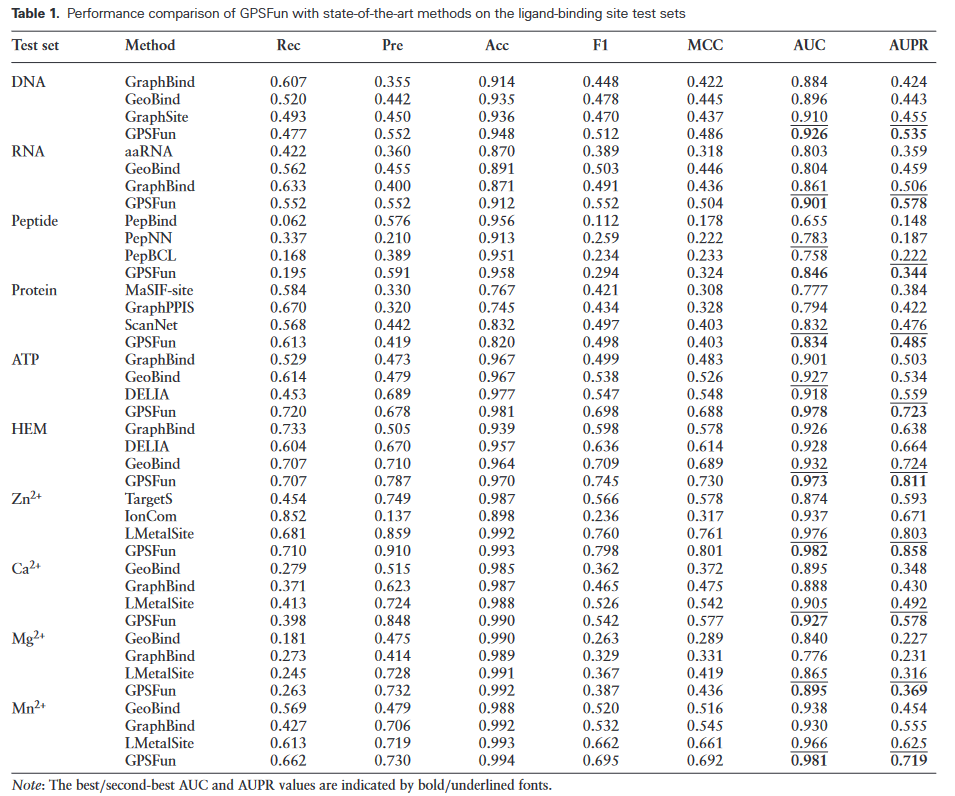
表 1
对于蛋白质-配体结合位点的预测,作者将GPSFun与最新的基于序列的方法进行了比较,包括GraphSite、PepBind、PepBCL、TargetS和LMetalSite,以及基于实验结构的方法,包括GraphBind、GeoBind、aaRNA、PepNN、MaSIF-site、GraphPPIS、ScanNet、DELIA和IonCom。如表1所示,GPSFun在DNA、RNA、肽、蛋白质、ATP、HEM、Zn2+、Ca2+、Mg2+和Mn2+的独立测试集中,AUPR分别超过所有竞争方法17.6%、14.2%、55.0%、1.9%、29.3%、12.0%、6.8%、17.5%、16.8%和15.0%。为了进一步说明语言模型的序列嵌入和预测结构的有效性,作者进行了消融研究。通过使用ProtTrans嵌入作为序列特征,而不是之前使用的MSA配置文件,在十种配体中的平均AUPR增加了4.2%。另一方面,去除结构信息导致平均AUPR大幅下降19.3%。此外,去除GPSFun中的几何特征化工具也导致平均AUPR显著下降11.5%,这突显了GPSFun感知蛋白质几何结构的重要性。
GPSFun在GO预测上的性能
对于GO预测,GPSFun在测试集的分子功能(MF)、生物过程(BP)和细胞成分(CC)上,AUPR分别比基于序列的方法BLAST-KNN、DeepGOPlus和GOLabeler,基于预测结构的方法Foldseek-KNN,以及基于蛋白质-蛋白质相互作用网络的方法DeepGraphGO和NetGO高出11.6%、25.3%和5.8%以上。此外,GPSFun的表现与作者之前的SPROF-GO工具相当。GPSFun还很好地适用于非同源蛋白质。
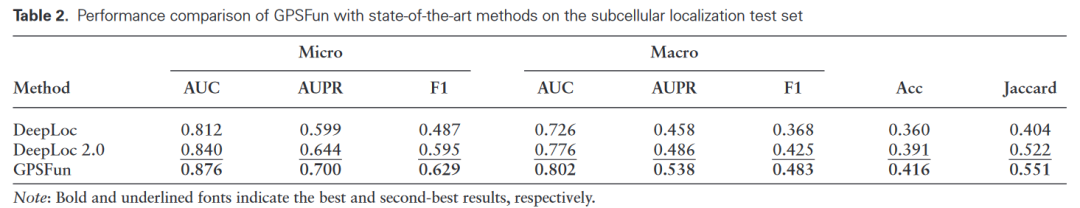
表 2
在亚细胞定位预测方面,GPSFun的微观和宏观AUPR分别比基于序列的预测器DeepLoc和DeepLoc 2.0高出8.7%和10.7%以上(见表2)。与BLAST-KNN、Foldseek-KNN和不含结构信息的基线模型相比,GPSFun也表现出更好的性能。
编译 | 黄海涛
审稿 | 曾全晨
参考资料
Yuan, Q., Tian, C., Song, Y., Ou, P., Zhu, M., Zhao, H., & Yang, Y. (2024). GPSFun: geometry-aware protein sequence function predictions with language models. Nucleic Acids Research, gkae381.














腾讯云开发者
